Tom recently emailed me this question: Do you know how to find out how many of the compounds that appear in the chemical literature are mentioned just once? Intrigued, I first set out to find out how many substances, as Chemical Abstracts refers to the them, there were as of 5 June, 2025. There is a static estimate here (219 million), but to get the most up to date information, I asked CAS directly. They responded immediately (thanks Lee!) with 294,778,693 on the date mentioned above. It is not actually possible to answer the first question itself using CAS SciFinder, but again CAS came up with a value: “there are 113,383,649 substances in CAS Registry with only one CAplus citation” equivalent to “38.5% of the current substances have only 1 reference.” I should add this estimate was qualified by “that can be misleading, since that includes salts, multicomponents, etc. But that’s a first pass.” I am actually impressed that as many as 61.5% are mentioned more than once, since before learning the answer, I had intuitively guessed that percentage as being much lower.
My mind then went back to the year 1974, when my PhD thesis was published.[cite]10.14469/spiral/20860[/cite] As part of this research, I had managed to synthesize several sterically hindered indoles, culminating in the preparations of 2-Methyl-3,5-di-t-butylindole (3, R=Me)and 2,4,6-tri-t-butylindole (3, R=t-Butyl) by the route shown below (R= Me, t-Butyl – a different route also gave the same product). I was very proud of this, since my research supervisor intimated to me a few years later that he had not believed I would succeed, on the grounds that making sterically hindered systems can be quite challenging! This work was published in a journal in 1975.[cite]10.1039/P29750001209[/cite]

Next, to find out what “impact” this work has had in the intervening 50 years. Well, a CAS SciFinder search revealed that 2-Methyl-3,5-di-t-butylindole (3, R=Me) was one of the 38.5% of the current substances that have only 1 reference, to just our own work. Zero impact then! But worse was to come – 2,4,6-tri-t-butylindole (3, R=t-Butyl) did not even have 1 reference – as far as CAS was concerned, it was an unknown compound! So too were the precursors 2-methyl-3,5-di-t-butylaniline (1) and the anilides 2 (R=Me, t-butyl).
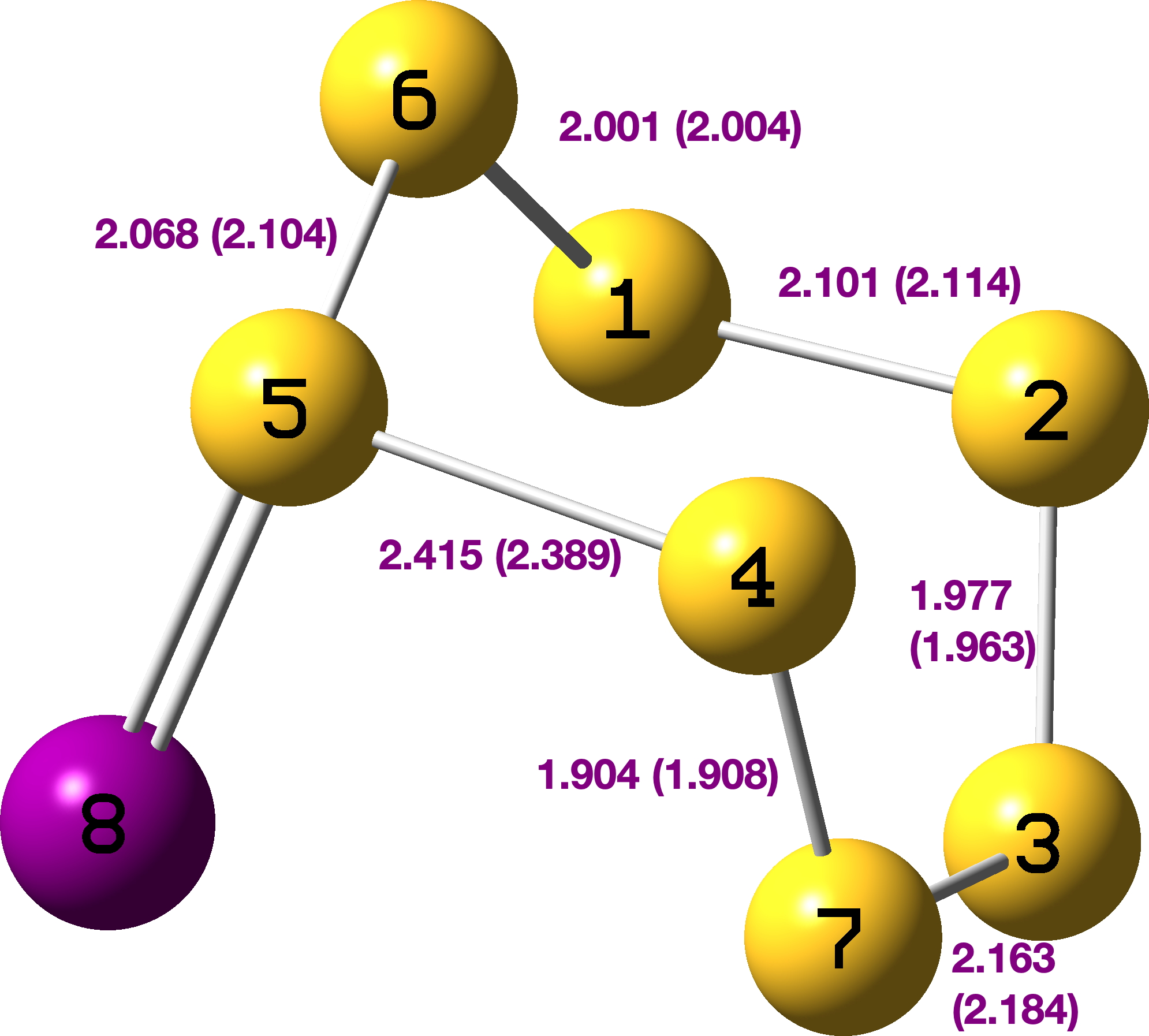
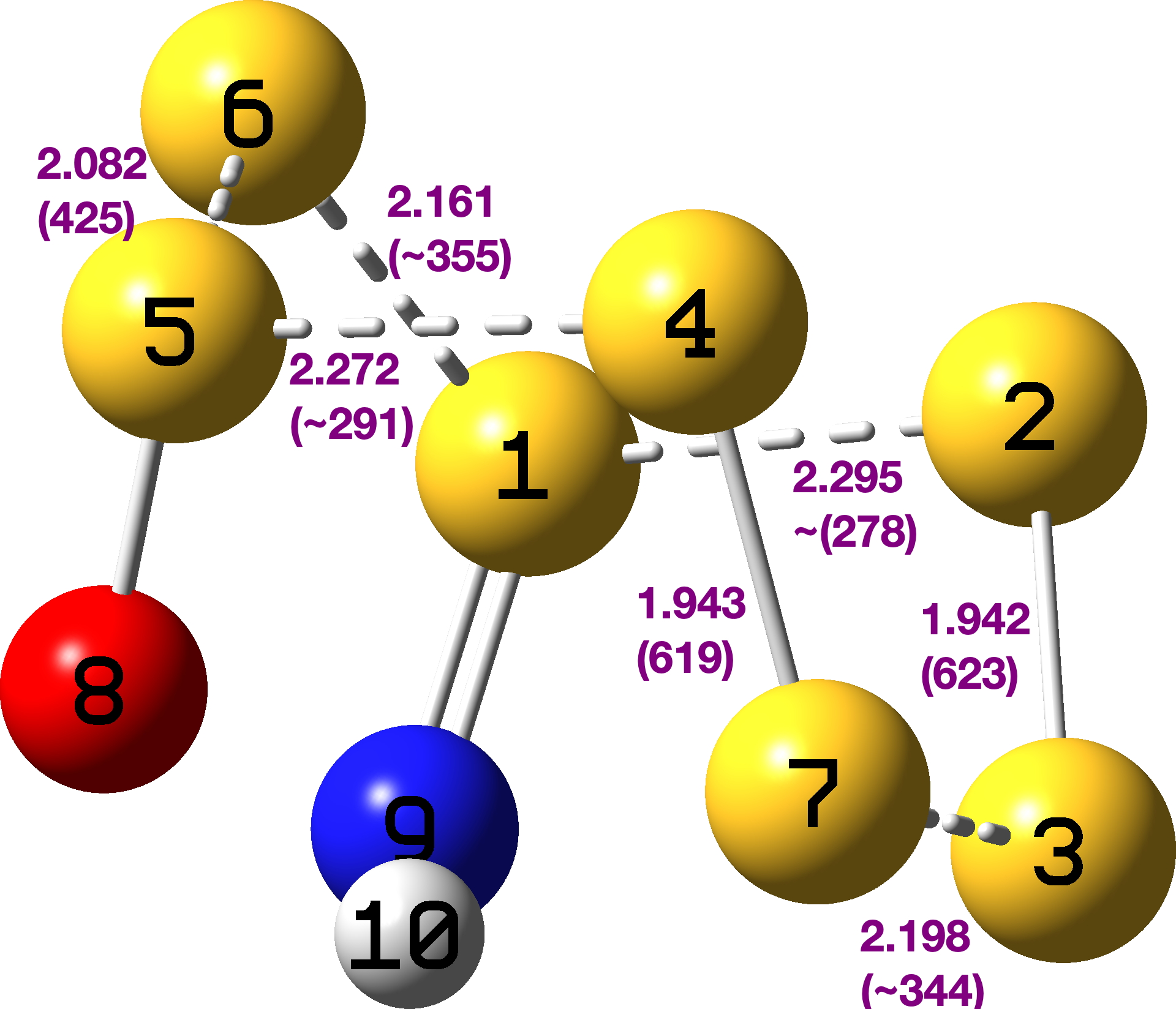
The explanation can be found – at least in part – by reading our article[cite]10.1039/P29750001209[/cite] and from the computational modelling I did some forty years later.[cite]10.59350/1jhn9-9v717[/cite] We were measuring kinetic isotope effects on the rate of diazo-coupling of these indoles and had noted in the article that 2,4,6-tri-t-butylindole was so hindered it simply did not diazo-couple at any measurable rate. As a result, it was not included by us in the experimental section detailing its synthesis (we really should have). The absence of the anilides 2 in the CAS database is perhaps understandable, since they are merely precursors to the final cyclisation and these are not always characterised as fully as final products. I have retrieved the experimental information in my PhD thesis[cite]10.14469/spiral/20860[/cite] and reproduce it here so that you can see it as well. I note that the anilide 2, R=Me) is mentioned only in passing (red text below) whilst for 2, R=t-Butyl, only an m.p. and mass spec weight are included.
I have now set myself the challenge of whether substances 1 and especially 3 (R=t-Butyl) at least can be retrospectively added to the CAS database. Watch this space!
2-Methyl-3,5-di-t-butylaniline.
Bromine (8g) was added to dimethylsulfide (3.2g) in dichloromethane (40 ml) at -46° (chorobenzene/N2 cooling bath) with no precautions taken to exclude moisture. A yellow crystalline precipitate of bromosulfonium bromide salt was formed. 3,5-Di-t-butyl aniline (10g) and triethylamine (5g) in dichoromethane (10 ml) were added dropwise, during the course of which the yellow salt dissolved and white crystals of triethylammonium bromide were deposited. After 2 hours at -46°, a solution of sodium (2.5g) in methanol (15 ml) was added, resulting in the production of a white precipitate of sodium bromide. After 8 hours at 20° the rearrangement was essentially complete and the solution was shaken with water, the solvent separated and evaporated to give a yellow oil (12g, 95%) which crystallised on standing. δ 1.30 (9H, s), 1.47 (9H, s) 2.13 (3H, s), 4.12 (4H, br), 6.53, 6.83 (2H, dd, JAB 2Hz). m/e 265 (M+), 218 (M+-CH3S+).
Raney nickel (prepared from 210g of 50% Na/Al alloy) was stirred with a solution of the 2-methylthiomethyl-3,5-di-t-butylaniline (32g) in ethanol (150 ml) at 70° for 1 hour. Filtration and evaporation of the solvent gave an oil which on distillation gave 2-methyl-3,5-di-t-butylaniline (66%), b.p. 126°/2.7 mm. δ 1.25, 1.38 (18H, d), 2.17 (3H, s), 3.27 (2H, s), 6.43, 6.75 (2H, dd, JAB 2Hz).
2-Methyl-3,5-di-t-butylindole.
2-Methyl-3,5-di-t-butylaniline (2g) in ether (20 ml) and triethylamine (1g) was mixed with acetyl chloride (1.2 g) in ether. After 1 hour the ether was washed with 0.01N HCl and the solvent removed to give the acetyl derivative (90%). The acetyl derivative was cyclised by potassium t-butoxide at 360° to give a melt which was boiled up with water. Ether extraction followed by crystallisation from hexane gave 2-methyl-4,6-di-t-butylindole (30%), m.p. 176°. νmax 3370, 1617, 1538, 849, 784, 755 cm-1. δ 1.35, 1.45 (18H, d), 2.37 (3H, s), 6.27 (1H, m), 6.97 (1H, s), 7.4 (1H, br, exchanges with D2O). λmax (log ε) 223 (4.35), 272 (3.95). m/e 243 (M+), 225 (M+-15). Found C, 81.95; H, 11.41; N, 6.19%. C15H25N requires C, 82.12; H, 11.48; N 6.38%.
2,4,6-Tri-t-butyl indole.
2-Methyl-3,5-di-t-butyl aniline was acylated with trimethyl acetyl chloride in ether to give the anilide (97%), m.p. (ether) 215°, m/e 303 (M+). Fusion with potassium t-butoxide at 350C gave on cooling a solid which was treated with water, giving brown crystals of the 1:1 t-butanol complex. These were dried and sublimed very slowly at 70° to give a colourless glass (25%), pure by nmr and tlc. νmax 3450, 3310, 2960, 2870, 1645, 1600, 1370, 800 cm-1. δ 1.30, 1.35, 1.48 (27H, t), 6.25 (1H, d, 2Hz), 6.95 (1H, d, 2Hz, 7.72 (1H, s, exchanges with D2O). m/e 285 (M+), 270 (M+-15). Found C, 84.12; H, 10.97; N, 4.76%. C20H31N requires C, 84.14; H, 10.94; N 4.90%.