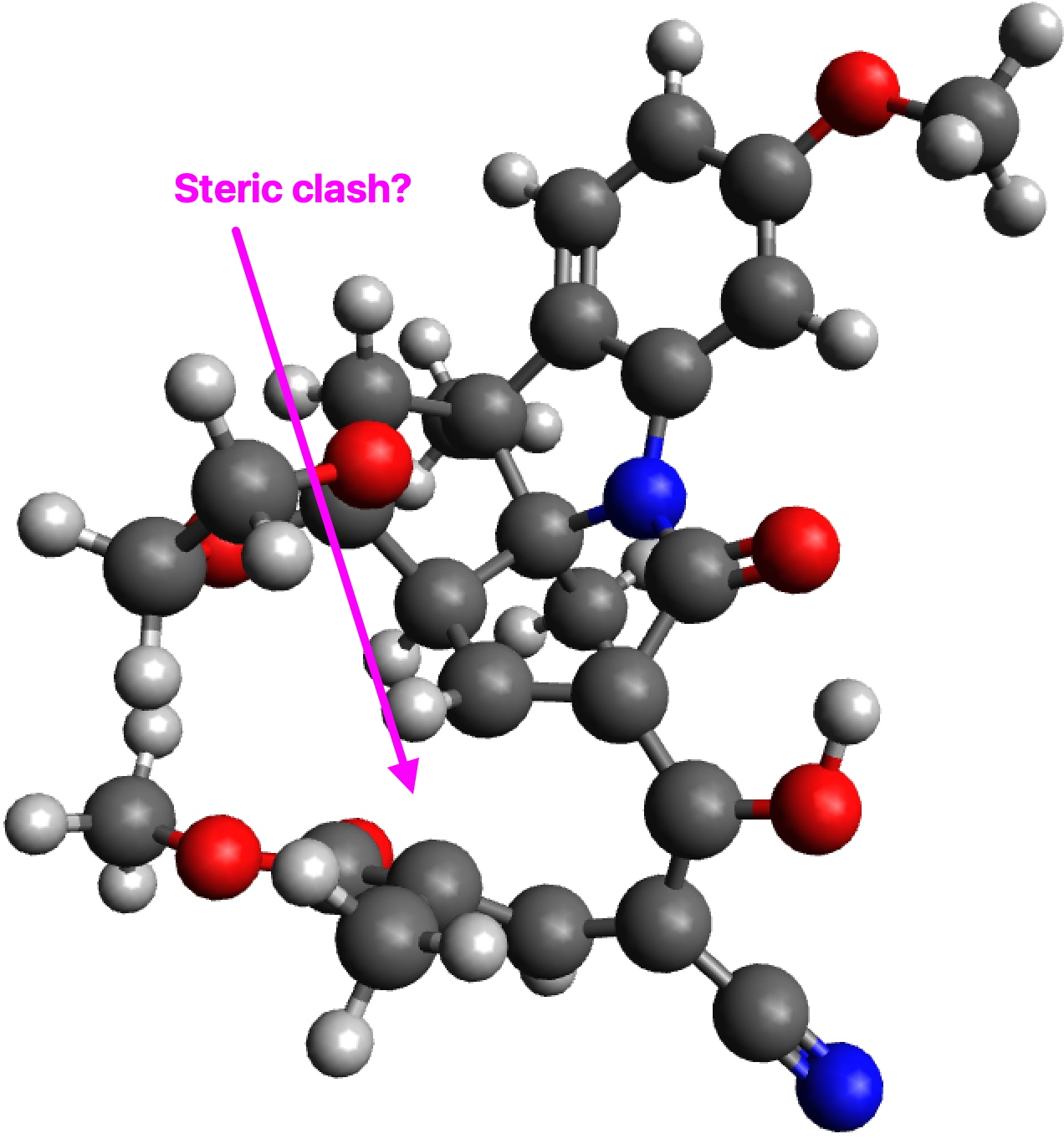
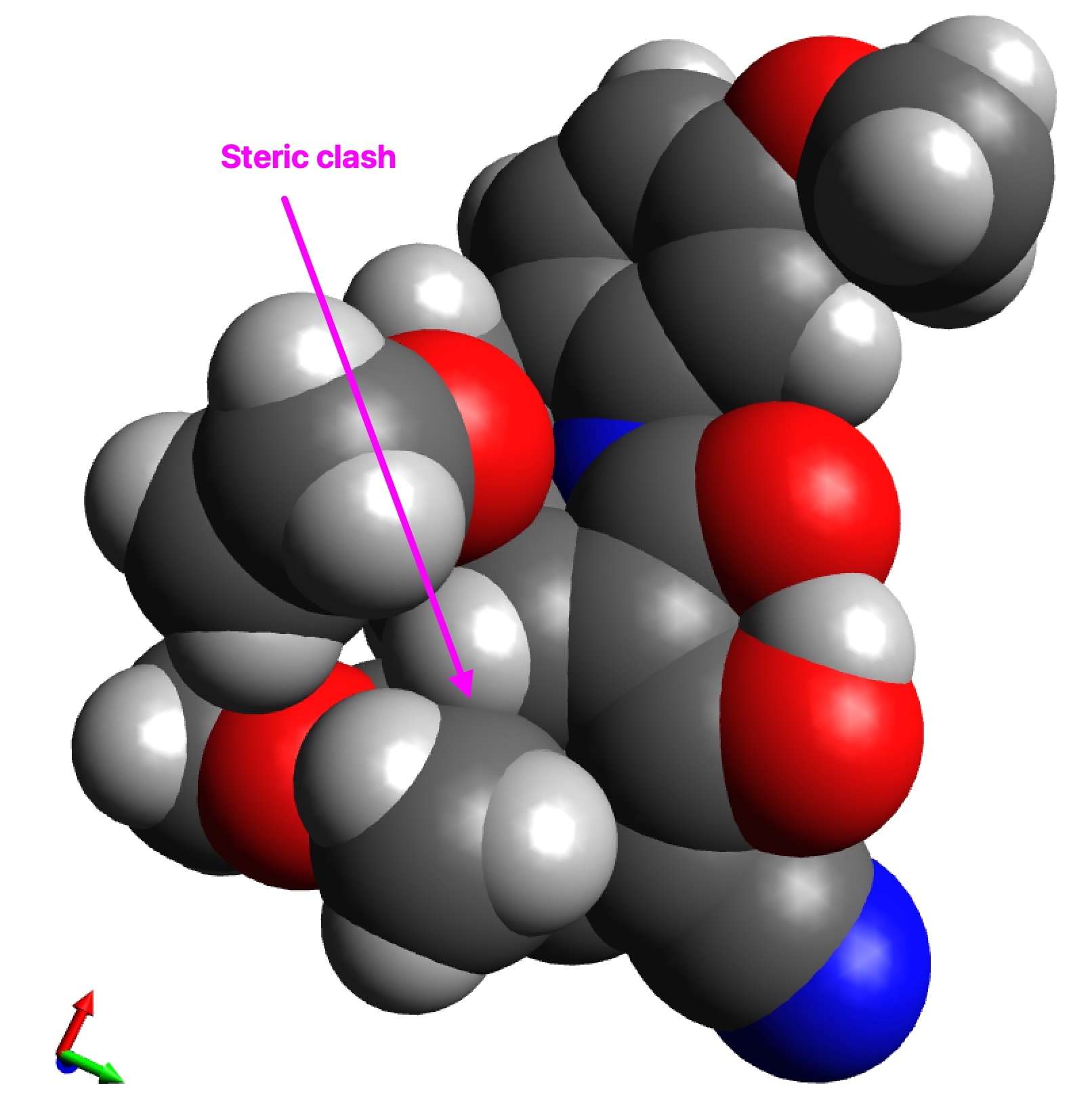
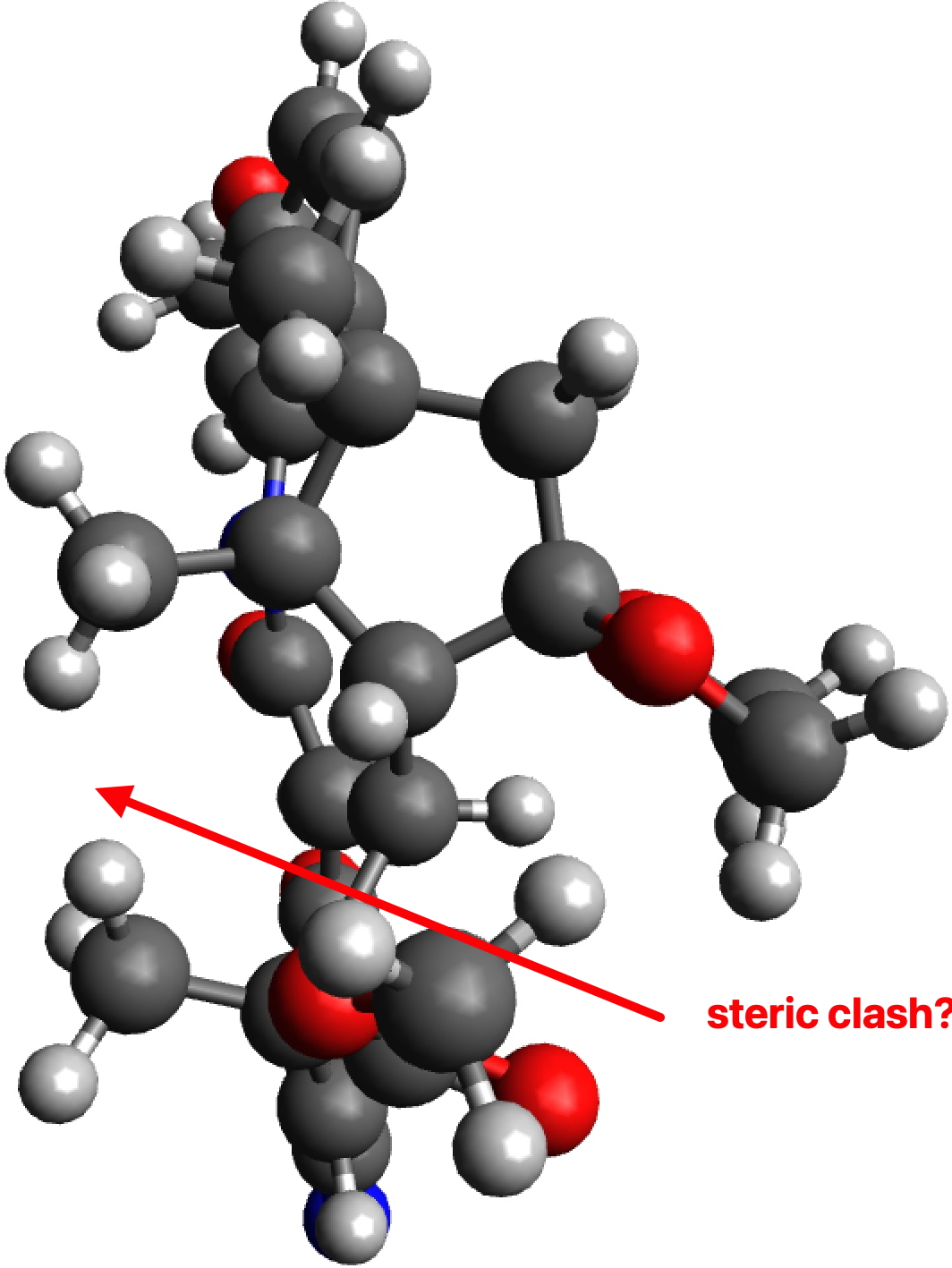
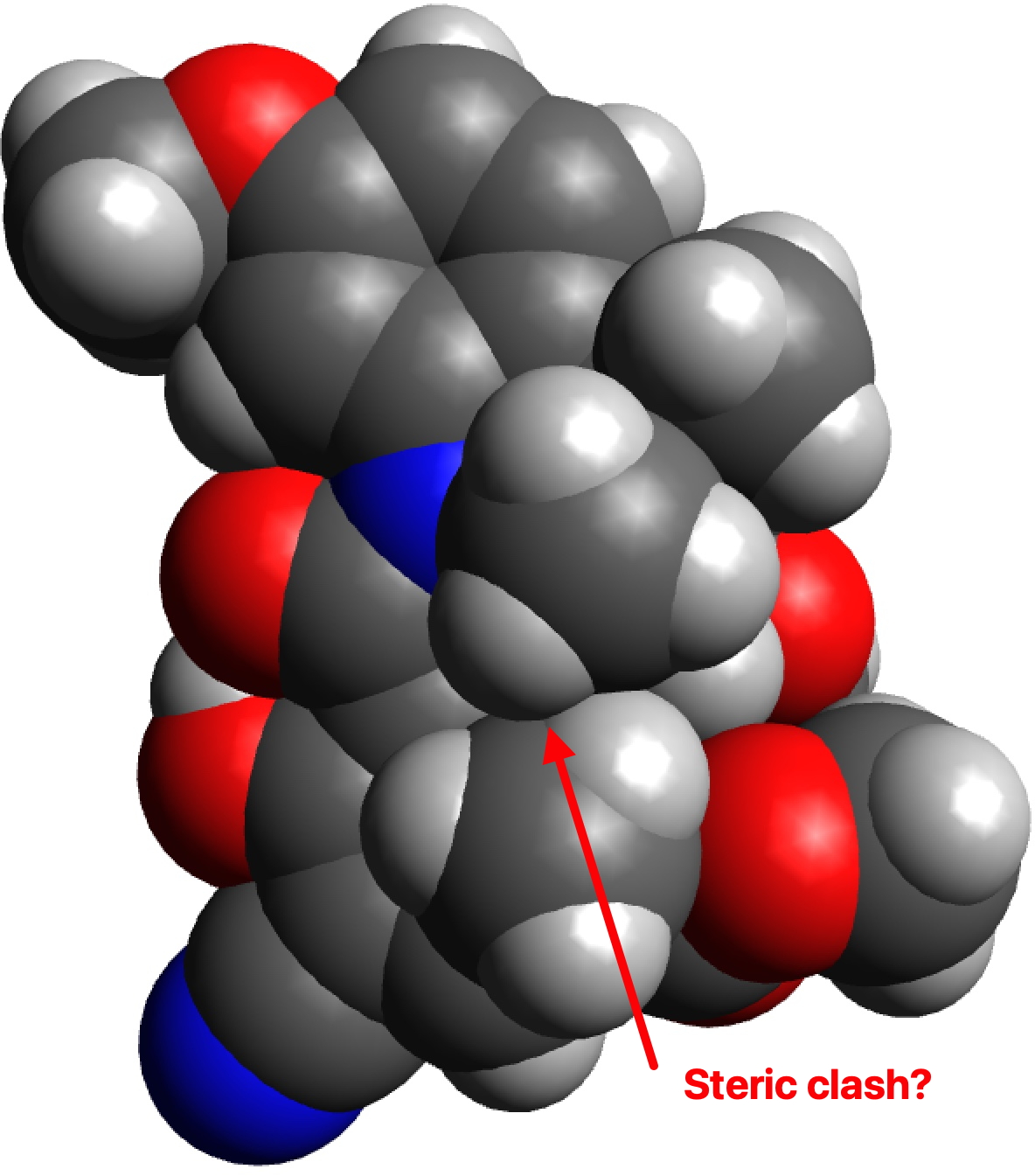
Whilst I was discussing the future of scientific publication in the last post, a debate was happening behind the scenes regarding the small molecule cyclopropenylidene. This is the smallest known molecule displaying π-aromaticity, but its high reactivity means that it is unlikely to be isolated in the condensed phase. A question in the discussion asked if substituting it with a large sterically hindering group such as R=Et3C might help prevent its dimerisation and hence allow for isolation of the monomer so that its properties can be studied.
 |
 |
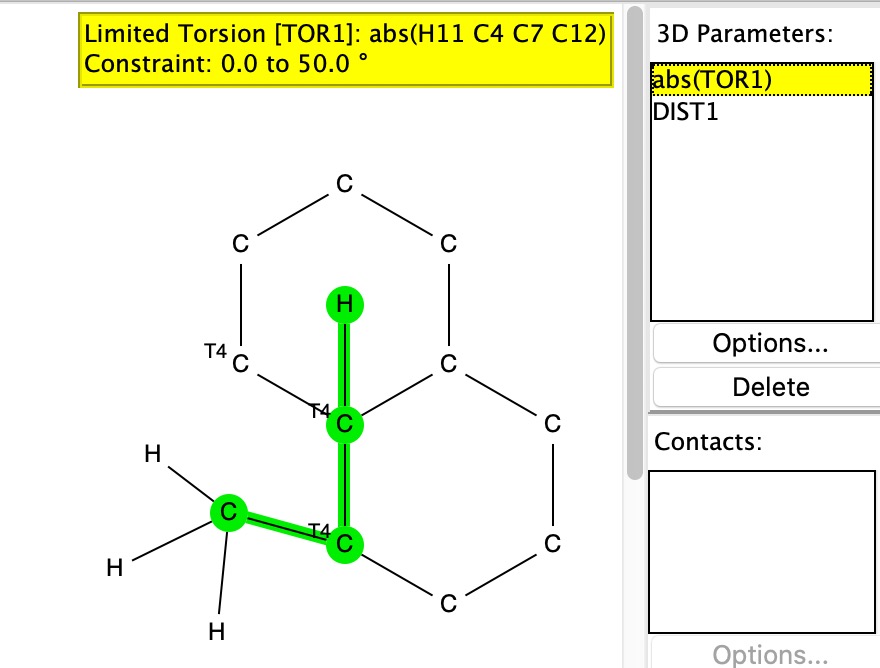
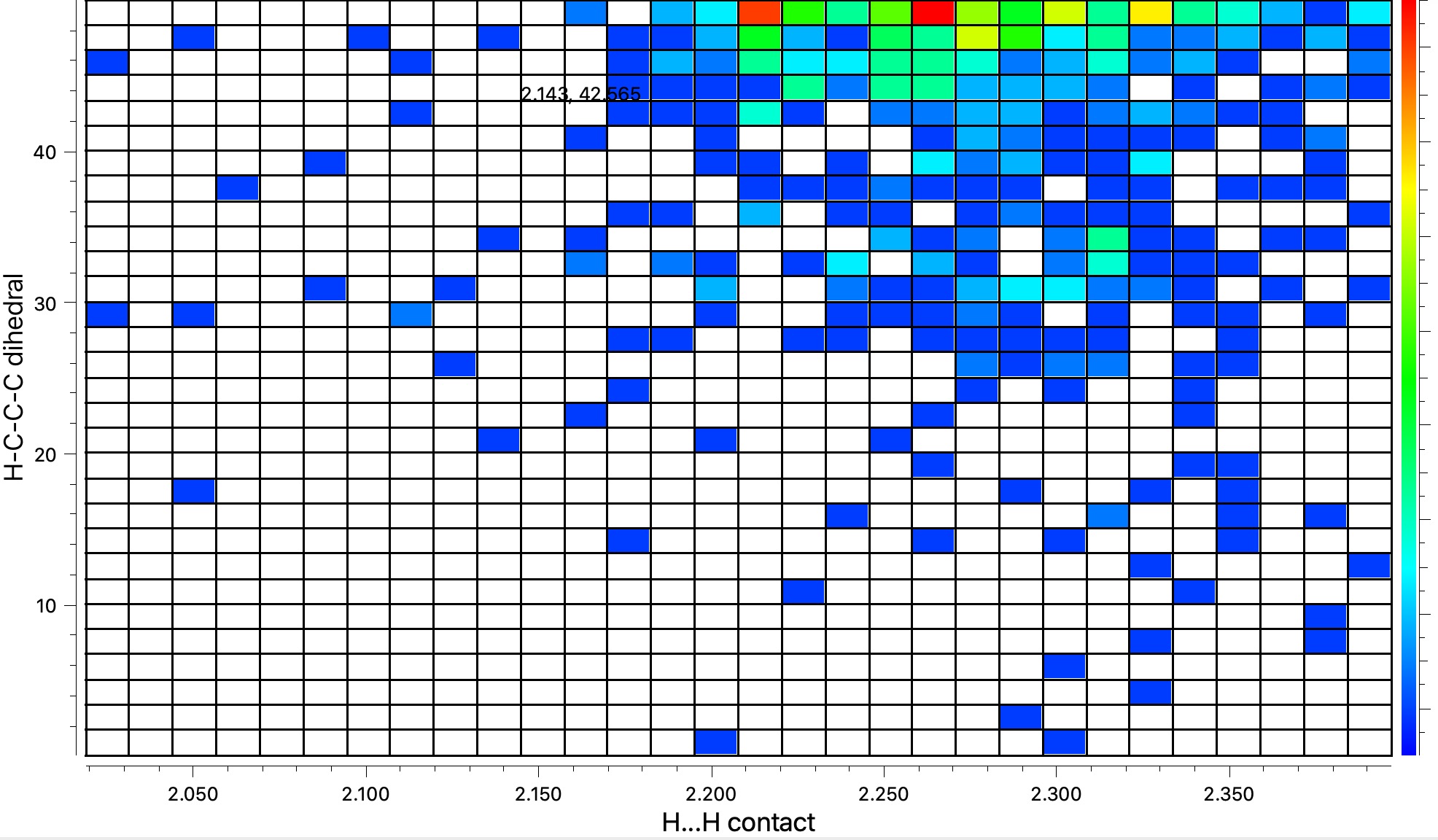
But first, a crystal structure search for this interesting group, Et3C, which is one step up in steric size from the very much better known Me3C or t-butyl. As it happens 34 examples emerge, and the dihedral angle distribution of the three ethyl groups is shown below. The three clusters all correspond to conformations with two gauche and one anti ethyl group. 
Whilst on the topic of crystal structures, I note that there are 5 examples known of the next steric homologue, i-Pr3C and a surprising 18 of t-Bu3C. I will discuss these groups elsewhere.
Next, a protocol for modelling the dimerisation: ωB97XD/Def2-SVPP/SCRF=dichloromethane. The IRC for R=H is shown at DOI: 10.14469/hpc/8705 and here I show that for R=Me3 showing a slightly larger barrier.

The results for three substituents are summarised in the table below which show that the barrier is a maximum for the t-butyl group and then decreases slightly for the apparently “larger” Et3C group.
| R | ΔG‡ | FAIR Data DOI |
|---|---|---|
| H | 14.4 | 10.14469/hpc/8470 10.14469/hpc/8495 |
| Me3C | 16.0 | 10.14469/hpc/8706 10.14469/hpc/8707 |
| Et3C | 15.4 | 10.14469/hpc/8712 10.14469/hpc/8724 |
| iPr3C* | 25.5 | 10.14469/hpc/8722 |
| tBu3C* | 101.7 | 10.14469/hpc/8771 10.14469/hpc/8743 |
The analysis of this result is as noted in the discussion alluded to above, which is that these large groups, bristling with exposed hydrogen atoms, are strong dispersion attractors, at the right interatomic distances. The t-butyl group must be slightly sterically repulsive for the dimerisation reaction, but those dispersion attractions stabilise the slightly larger Et3C group. This could be tested further with R=i-Pr3C and t-Bu3C*.
I wanted to end this by going back to the opening line of this post. It struck me that the three posts here on the topic of cyclopropenylidene and the discussion they induced is not dissimilar from the “octopus” publishing modelling I had previously looked at.
- It started with setting out the initial seeding publication, in this case by noting that cyclopropenylidene had recently been reported in the atmosphere of Saturn’s moon Titan.[cite]10.3847/1538-3881/abb679[/cite].
- The hypothesis was that this molecule might be π-aromatic, an observation not noted in the original report (DOI: 10.14469/hpc/8716)
- A protocol for testing this hypothesis was to look at the occupied molecular orbitals of this molecule using a DFT-based quantum method (DOI: 10.14469/hpc/8716)
- The data resulting from this protocol is published (DOI: 10.14469/hpc/8714).
- Visual analysis showed two π-electrons (4n+2, n=0) i0n a molecular orbital fully delocalised around the three membered ring, which itself implies charge asymmetry in the molecule (DOI: 10.14469/hpc/8716)
- The original hypothesis of ring aromaticity was thus confirmed.
- A real-world problem then arose in the discussion relating to the dipole moment of this species resulting from the charge asymmetry.
- The review in this case was by comments posted to the blog posts here (a form of non-anonymous review).
- These reviews then spawned a new hypothesis, that a molecule based on cyclopropenylidene might support a record-large dipole moment (DOI: 10.14469/hpc/8717)
- This idea started a new cycle in which cyclopropenylidene might react with a source of dicarbon to give the desired molecule (DOI: 10.14469/hpc/8717)
- This cycle in turn spawned the current discussion, which relates to whether cyclopropenylidene might have a sufficiently long bimolecular lifetime to react with another molecule in preference to reacting with itself (DOI: 10.14469/hpc/8715)
- With a fork into crystal structure mining of steric groups beyond t-butyl.
- The latter resulting in a further cycle likely to be started relating to the hypothesis of R = i-Pr3C as an interesting steric group.
So we see here what might map to three cycles of “octopus publishing”. Those cycles were however non-linear, in that they did not happen in quite the sequence outline above; the discussions forked and split out from the original cycle, re-entering at different points in the cycle. My point being that scientific research is indeed very often cyclical and non-linear, albeit traditionally its reporting taking place in a form where many of the individual aspects of this process are bundled together in the form of a research article, a box-set if you will, which you can binge on if you wish. The concept of Octopus publishing is to fragment this model into smaller, stand-alone episodes, linked perhaps by a metadata-based DOI crumb trail. Lets see if the perceived benefits of publishing in this way catch on in chemistry.
*Further entries added to table.