Mercury (IV) tetrafluoride attracted much interest when it was reported in 2007[cite]10.1002%2Fanie.200703710[/cite] as the first instance of the metal being induced to act as a proper transition element (utilising d-electrons for bonding) rather than a post-transition main group metal (utilising just s-electrons) for which the HgF2 dihalide would be more normal (“Is mercury now a transition element?”[cite]http://dx.doi.org/10.1021%2Fed085p1182[/cite]). Perhaps this is the modern equivalent of transmutation! Well, now we have new speculation about how to induce the same sort of behaviour for caesium; might it form CsF3 (at high pressures) rather than the CsF we would be more familiar with.[cite]10.1038/nchem.1754[/cite] Here I report some further calculations inspired by this report.
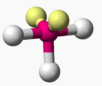
The argument goes something like this. Xenon difluoride (XeF2) is a well-known stable compound of xenon. Caesium comes immediately after xenon in the periodic table (electron shell properties [Xe].6s1) and so Cs+ would be iso-electronic with Xe. If the latter can form a stable difluoride (and higher), why not Cs? A neutral compound following this line of argument would therefore be CsF3.
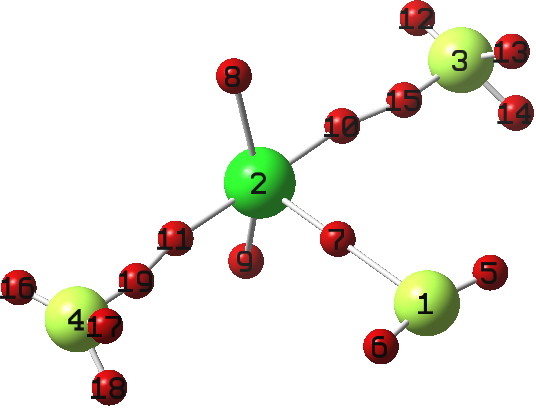
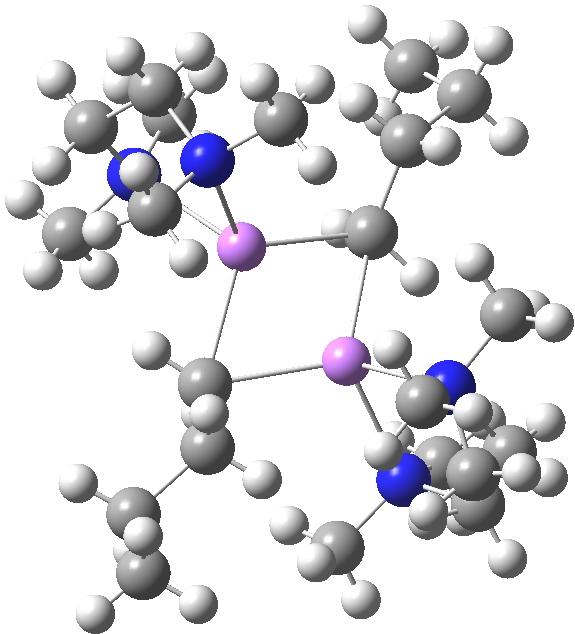
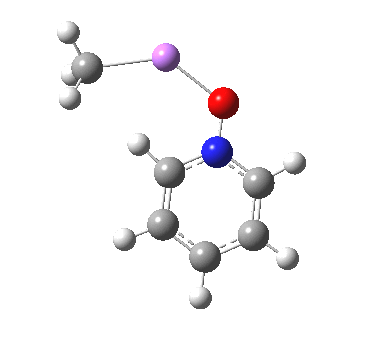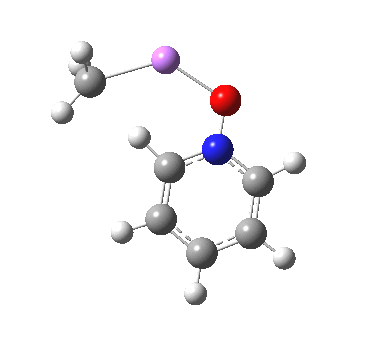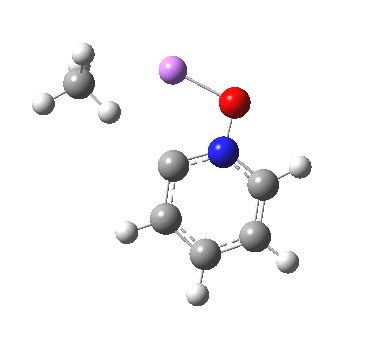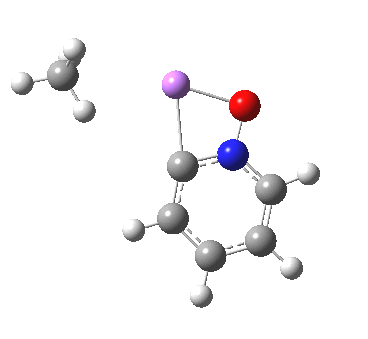
So here comes a calculation. I used a large basis set (Def2-QZVPPD basis for Cs), with a pseudopotential describing 46 core electrons (including the two d-shells) and a further 8 in the 5s/p shell to make up the [Xe] core + 1 extra in the 6s shell) using the ωB97XD functional to obtain the geometry, shown below.[cite]10.6084/m9.figshare.861029[/cite]

All 3N-6 normal vibrational modes are real, which indicates it is a proper minimum in the potential energy surface. A QTAIM analysis shows that ρ(r) at the bond critical points is pretty respectable for bonds (below). The QTAIM-derived average number of electrons on Cs is 7.4, which gives a charge of 1.6 on the Cs , thus involving the 5p shell as well as the 6s.

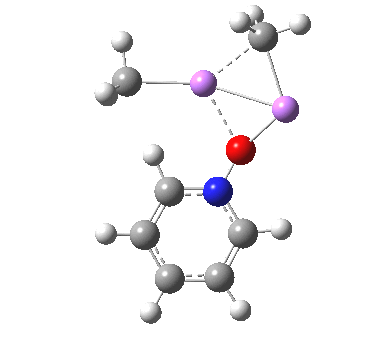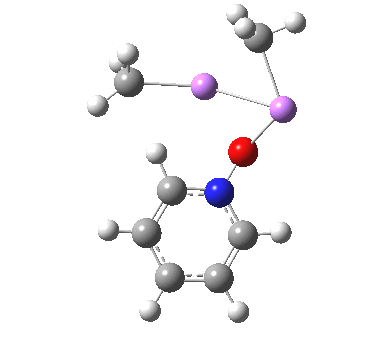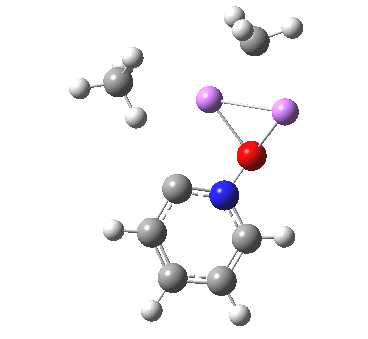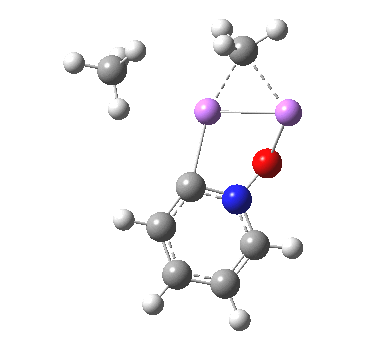
The most obvious question is what is the free energy change when the species dissociates to CsF and F2? This is exo-energic by 25.1 kcal/mol[cite]10.6084/m9.figshare.861030[/cite], not as large as you might have thought! Well, what about the barrier to such dissociation? The transition state for this process is shown below, delightfully asymmetric![cite]http://hdl.handle.net/10042/26513[/cite] and this gives a free energy barrier of 33.9 kcal/mol (the IRC smoothly defines this reaction[cite]10.6084/m9.figshare.861038[/cite],[cite]10.6084/m9.figshare.861047[/cite]).

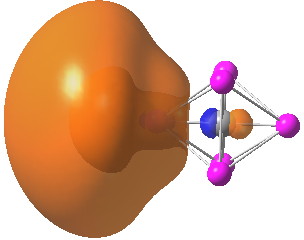
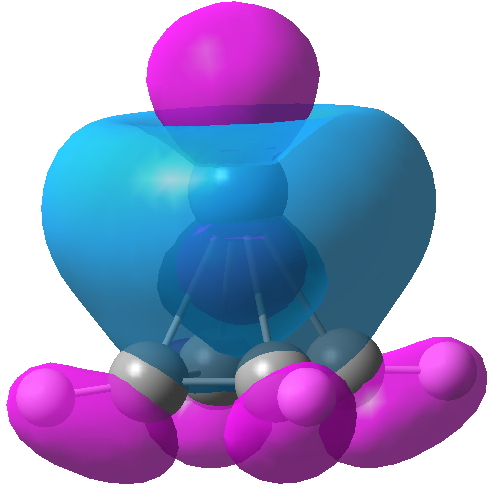
We may conclude from this brief foray that in CsF3 caesium would indeed deserve to be called a p-block element, although this is not quite as good as it sounds. Take a look for example at the highest occupied molecular orbital. It certainly involves the p-block, but this orbital is in fact anti-bonding, and the Cs-F bonds derive their strength from the lower occupied orbitals.


And the final take-home message. The report of this molecule[cite]10.1038/nchem.1754[/cite] suggests it could be stable under high pressure. Here, the free energy barrier to dissociation is calculated to be indeed high, which implies that if made it could be kinetically quite stable even under normal pressures (in an inert matrix where it would be prevented from reacting with itself).
POSTSCRIPT: The LUMO below is also antibonding, but is mostly 6s on Cs. If one force-populates this by a double excitation from the HOMO, the resulting state is higher in energy (the wavefunction is stable to both singlet and triplet excitations). Which shows that Cs utilises (antibonding) 5p rather than (antibonding) 6s in this species.