In the previous post,[cite]10.59350/jhsbq-sfs70[/cite] I noted the photochemical isomerisation of a pyrimidone into what is called the bicyclic Dewar form, being part of a solar energy storage system.[cite]10.1126/science.aec6413[/cite] A colleague (thanks Alan!) has recollected a very similar example dating from 1965[cite]10.1039/C19650000468[/cite] in which a related molecule known as a diazepinone 72 (scheme below) is converted by light into a Dewar form 73.
This example was first highlighted in Woodward and Hoffmann’s (WH) famous 1971 book on the topic of the conservation of orbital symmetry† in which they noted that the Dewar form of a diazepinone (73 in scheme) had been observed[cite]10.1039/C19650000468[/cite] to thermally “revert to diazepinone in the dark”.† The original authors[cite]10.1039/C19650000468[/cite] also specifically noted that the Dewar diazepinone was “stable to storage” after being protonated. These two properties are the exact inverse of the recent report,[cite]10.1126/science.aec6413[/cite] whereby the photochemical bicyclic form of pyrimidone was found to be thermally stable, but very rapid ring opening was induced by protonation with acid. Here I explore whether these apparently contradictory reports can be reconciled.

In discussing the reaction of 72 in their book‡, WH suggest that the stereochemical aspects of the thermal ring opening of 73 could be explained using their rules by prior inversion of the ring nitrogen stereochemistry to that of 73-inv, followed by conrotatory/antarafacial ring opening to 72. Here, with the help of ωB97XD/Def2-TZVPP/DCM DFT calculations,[cite]10.14469/hpc/15948[/cite] I discuss whether this suggestion is viable, and also propose an alternative mechanism (72-trans, Scheme above).
Firstly, I show the calculated reaction path[cite]10.14469/hpc/15975[/cite],[cite]10.5281/zenodo.20455914[/cite] along which HTS3 and 73-inv are found, being the WH suggestion for this reaction.
![]()

Figure 1. IRC Energy plot and animation for TS3
- At IRC ~8, (Figure 1) the potential shows what can be called a “hidden transition state”, at which point the gradient norm is close to zero. This is the point labelled HTS3, followed soon after by a “hidden intermediate” (IRC ~4) or 73-inv. The process corresponds to inversion of the nitrogen lone pair to produce a bicyclic species with a trans ring fusion. These are both “hidden” because the gradient norm (Figure 2) does not actually reach a value of 0.0 as required for “real” transition states and intermediates, but comes very close.
- ΔG‡298 at these points relative to the starting point is ~34 kcal/mol, rather higher than would be needed for a truly thermal reaction. The CN bond length has not yet started to change (Figure 3).
- At IRC = 0.0 the true transition state is reached (TS3), involving WH-allowed antarafacial cleavage (Figure 5) of the bicyclic C-N bond (length @TS 2.035Å). The energy is now ~65 kcal/mol above the starting point, which makes this pathway very unlikely.
- The thermal reaction is exothermic by -19 kcal/mol (Figure 1), significantly less than that for Dewar pyrimidone.
![]()
Figure 2. Gradient norm plot for TS3
![]()
Figure 3. C-N bond length plot for TS3
![]()
Figure 4. Dipole moment plot for TS3, just for fun!

Figure 5. Geometry of TS3, showing C-N bond with antarafacial component (top face connecting bottom face) corresponding to conrotation (both clockwise) of the two termini.
Next, I tried an alternative mechanism, involving direct ring opening via TS1 to give a 7-ring with a trans bond, 72-trans. [cite]10.14469/hpc/15960[/cite],[cite]10.5281/zenodo.20279953[/cite] (Figure 6). Back in 1971, 7-rings with trans bonds were a rarity, so WH were probably reluctant to suggest this.

Figure 6. Energy plot for TS1
- The activation energy (corresponding to ΔG‡298 27.84 kcal/mol) is looking much better, matching to a slow (hours, days) thermal reaction at room temperatures. This value is somewhat less than the value of 32.9 kcal/mol for the analogous ring opening of Dewar pyrimidone,[cite]10.59350/jhsbq-sfs70[/cite] probably because the larger 5-ring ring means less transition state strain.
- The reaction again occurs with conrotation/antarafacial (Figure 7), C-N 2.192Å.
- But it is now endothermic by about +15 kcal/mol, reflecting the relatively high energy of a 7-ring product with a trans bond (Figure 6).

Figure 7. Geometry of TS1, showing C-N bond with antarafacial component (top face connecting bottom face) corresponding to conrotation (both clockwise) of the two termini.
To complete the mechanism, a route must now be found to convert 72-trans back to 72 itself.
- This can be done via a linear arrangement of the C-N-N atoms[cite]10.14469/hpc/15962[/cite] but the barrier to doing so is prohibitive (ΔG‡298 39.7 kcal/mol).
- An alternative is direct rotation about the C=N bond via an allylic biradical transition state (TS2),[cite]10.14469/hpc/15967[/cite],[cite]10.5281/zenodo.20406989[/cite] which yields ΔG‡298
26.74 kcal/mol. This value is less than that for TS1, and so is not rate determining.

Figure 8. Energy plot for TS2
When TS1 is protonated, ΔG‡298 becomes 26.4 kcal/mol (Figure 9, [cite]10.14469/hpc/15979[/cite],[cite]10.5281/zenodo.20474465[/cite], C-N 2.198Å) compared to the unprotonated value of ΔG‡298 27.8 kcal/mol. The slight decrease in barrier upon protonation does not match the observation[cite]10.1039/C19650000468[/cite] that protonated 73 is “stable to storage”. This still leaves open the question of why computations indicate that the rate of ring opening of Dewar diazepinone is relatively unchanged by protonation, whereas that of Dewar pyrimidone is greatly accelerated – the former involves protonating a hydrazine whereas the latter involves protonating an amide. Further models will need investigating to confirm whether this accounts for the essential difference in behaviour.

Figure 9. Energy plot for TS1 upon protonation.
To conclude, WH’s suggestion of a nitrogen inversion mechanism for the slow thermal pericyclic reaction of 72 followed by conrotatory C-N ring opening is instead replaced here by one invoking the electrocylic formation of a 7-ring intermediate with a trans bond and then biradical rotation of this bond.
‡ Woodward, R. B.; Hoffmann, Roald (1971). The Conservation of Orbital Symmetry (3rd printing, 1st ed.). Weinheim, BRD: Verlag Chemie GmbH (BRD) and Academic Press (USA). pp. 1–178. ISBN 978-1483256153. †The kinetics of this process were not noted, nor was the temperature.









































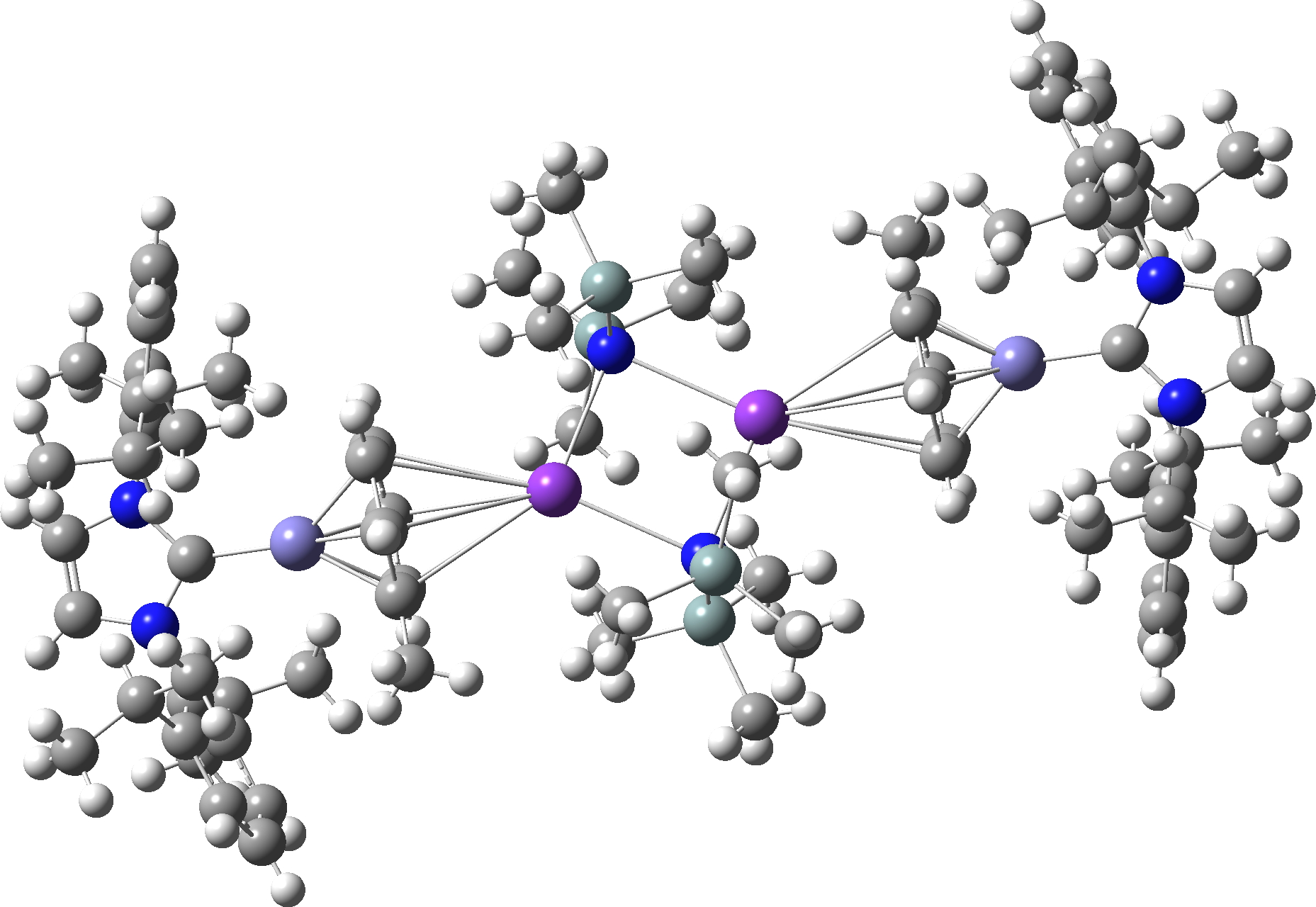







 Figure 1d. Calculated r2-SCAN-3c Raman activity for C58
Figure 1d. Calculated r2-SCAN-3c Raman activity for C58

