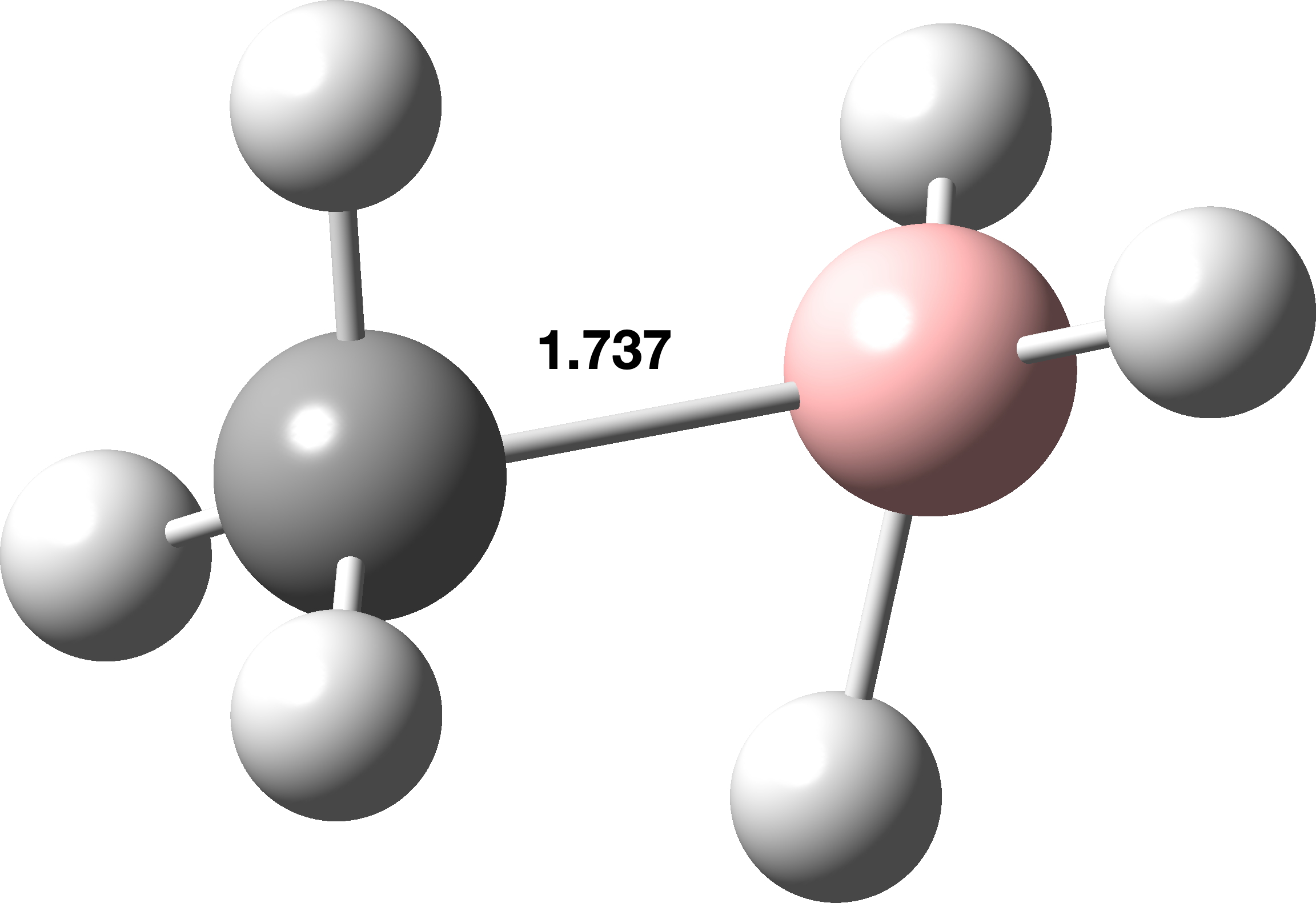
With AI and Machine learning needing data in abundance, interest in data discovery is intense. However, this type of discovery is somewhat different from more traditional data base searches, in that it is particularly suited for machine discovery as well as by humans. The discovery searches are conducted using an aggregated and federated metadata store, such as that curated by DataCite. How to construct a suitable search is however still not entirely human-friendly. The start point for understanding how to search is this resource: XML to JSON mappings and the XML referred to can be found here. [cite]10.14454/g8e5-6293[/cite] Since the learning curve to construct such data searches can be quite steep, I thought I would share as a library some recent searches I constructed for a talk I am giving. This post is essentially an extension and update of an earlier challenge I was set along these lines and which appeared here.[cite]10.1255/sew.2022.a10[/cite]
You can see that the searches come as components linked by Boolean operators, separated by strings such as +AND+, +OR+ or +NOT+. Essentially like a Lego constructor set, you can create your own searches by combining these components to suit your own needs. No doubt some AI-based procedure will come along that will convert natural language expressions of the intended search into the JSON-friendly strings you see below – at least that is the hope.
Part 1: Data discovery based on general properties such as the reporting Institution, the publisher or the Researcher
- Find all Data-related Works associated with Cambridge University and the American Chemical Society Publisher
- Find all Data-related Works associated with Imperial College and the American Chemical Society Publisher
- Find all Datasets OR Collections associated with Imperial College and the American Chemical Society Publisher and the term
Pyrazol in the Title or Description - Find all Datasets OR Collections associated with Imperial College and the American Chemical Society Publisher and the term
Pyrazol in the Title or Description and a specified Researcher - Find Datasets only associated with Imperial College and the term Pyrazol in the Title or Description
- Find just Datasets associated with a specific researcher
- Find Data-related Works associated with Cambridge University, the SubjectScheme FOS (Field of Science) and the Subject term *Chemical*
- Establish if a specified publication with a specified author has an associated FAIR Dataset or FAIR Collection:
- Establish how many journal publications by a specified author have an associated FAIR Dataset or FAIR Collection:
Part 2: Data discovery based on chemical properties such as NMR, IR or X-ray spectroscopy
- Find all Datasets associated with Chemical structure representation and NMR Media types,
NMR as a Subject and the title or description term
“Pyrazol” - Find all Datasets associated with Chemical structure representation and NMR Media types,
NMR Nuclei as a Subject, for 13C and the title or description term
“Pyrazol” - Find all Datasets associated with Chemical structure representation and NMR Media types,
NMR as a Subject, for HMBC Experiments and the title or description term
“Pyrazol” - Find all Datasets associated with Chemical structure representation and NMR Media types,
NMR as a Subject, using solvent “CD3OD” and the title or description term
“Pyrazol” - Find all Datasets associated with NMR Media types,
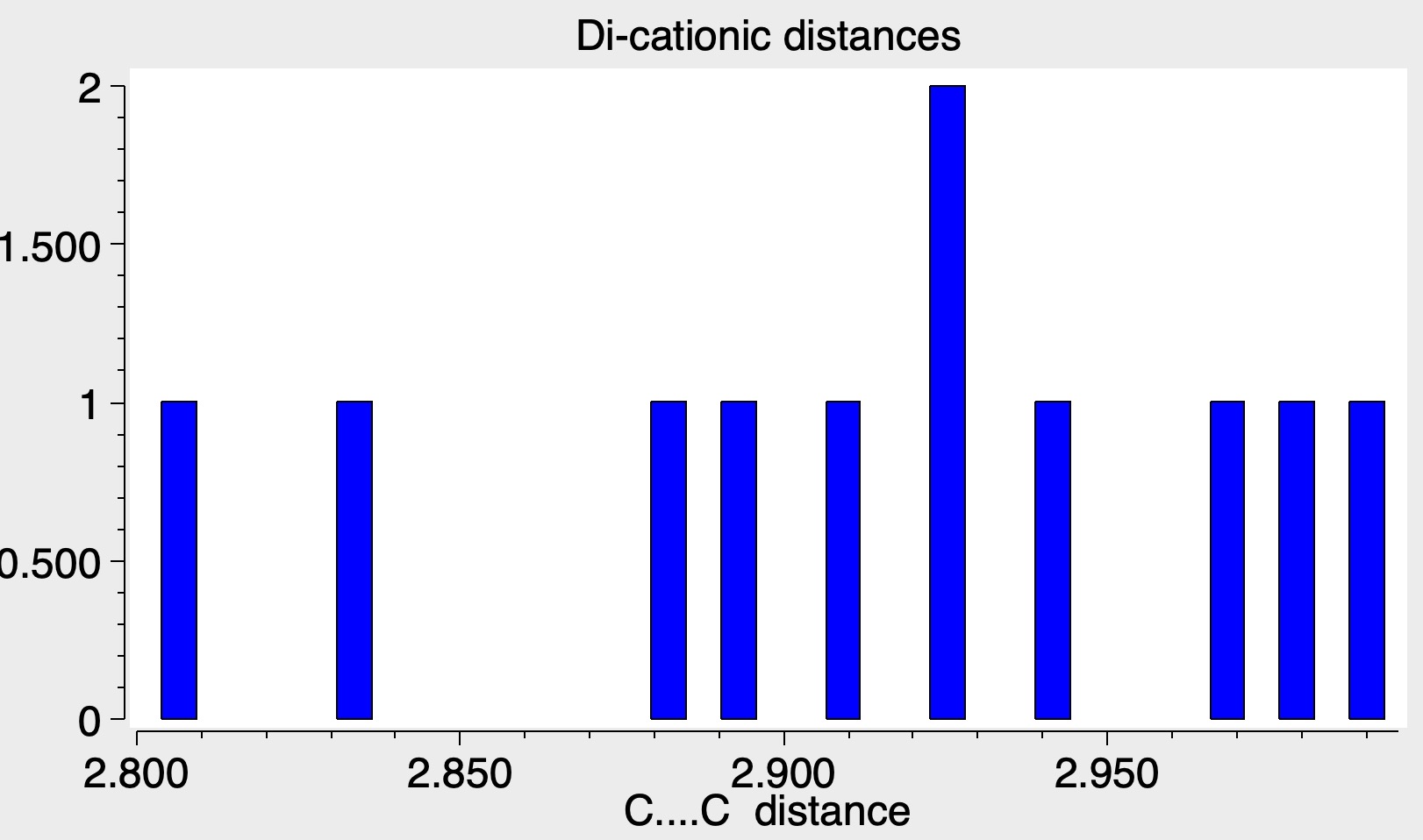
NMR as a Subject and InChIKey : OZEYXLXJQKVGCZ-UHFFFAOYSA-L - Find all Datasets associated with NMR Media types,
NMR as a Subject and the molecular formula component of the full InChI : InChI=1S/2C18H16N2O3.2C2H6O.Ca/c2*1-23-15-9-7-13 etc - Find all Datasets associated with Chemical structure representation Media types,
IR as a Subject and the title or description term
“Pyrazol” - Find all Datasets associated with a Chemical structure representation and Crystal structure
Media types, XRAY as a Subject and the
title or description term “Pyrazol”
Part 3: Data discovery based on chemical properties such as Computational modelling
- Find all Datasets associated with Chemical structure representation and Computation Media
types, COMP as a Subject and the title
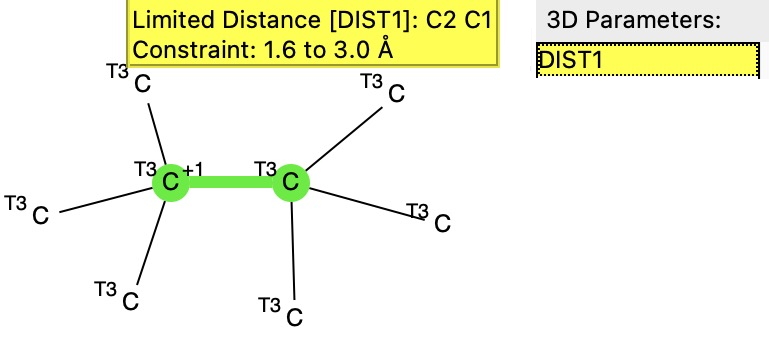
or description term “Pyrazol” - Find all Datasets associated with Computation Media types and the subject KIE for Hydrogen isotopes.
- Visual search:
?query=(media.media_type:chemical/x-gaussian-log+OR+media.media_type:chemical/x-gaussian-checkpoint)+AND+media.media_type:text/plain+AND+(titles.title:*Endo*+OR+descriptions.description:*Endo*+OR+titles.title:*Exo*+OR+descriptions.description:*Exo*)+AND+(subjects.subjectScheme:*KIE*)+AND+subjects.subject:1H/2H
17 datasets - API Search:
https://api.datacite.org/dois/?query=(media.media_type:chemical/x-gaussian-log+OR+media.media_type:chemical/x-gaussian-checkpoint)+AND+media.media_type:text/plain+AND+(titles.title:*Endo*+OR+descriptions.description:*Endo*+OR+titles.title:*Exo*+OR+descriptions.description:*Exo*)+AND+(subjects.subjectScheme:*KIE*)+AND+subjects.subject:1H/2H - Command line search:
curl https://api.datacite.org/dois/?query=(media.media_type:chemical/x-gaussian-log+OR+media.media_type:chemical/x-gaussian-checkpoint)+AND+media.media_type:text/plain+AND+(titles.title:*Endo*+OR+descriptions.description:*Endo*+OR+titles.title:*Exo*+OR+descriptions.description:*Exo*)+AND+(subjects.subjectScheme:*KIE*)+AND+subjects.subject:1H/2H
- Visual search:
One feature of this approach is that the searches themselves, which are across a globally aggregated metadata store, can change with time. So repeating some of the searches at defined time intervals can also give a dynamic indication of how a particular area of data is growing. Other searches are of course designed to give a single hit which probably will not change with time.
The above is based on an interpretation and implementation of the DataCite Schema, one which will eventually need to be agreed by the communities and sub-communities that might wish to use them. So beware, there may be other implementations covering similar data that would not eg be found by the above searches, particularly in the way the subject terms above are used. They are therefore included here purely to raise awareness of the potential that such an approach has – along with my observation that I had never attended any presentation where they have been discussed or shown. In the future, it seems likely that these JSON-based searches will themselves get automated and generated by software rather than by a human as here. When that comes, searching will never be the same again!
I also welcome suggestions for new search queries. This might either be accommodated using the existing metadata, or might require new additions to the metadata record. Please send them here as comments.