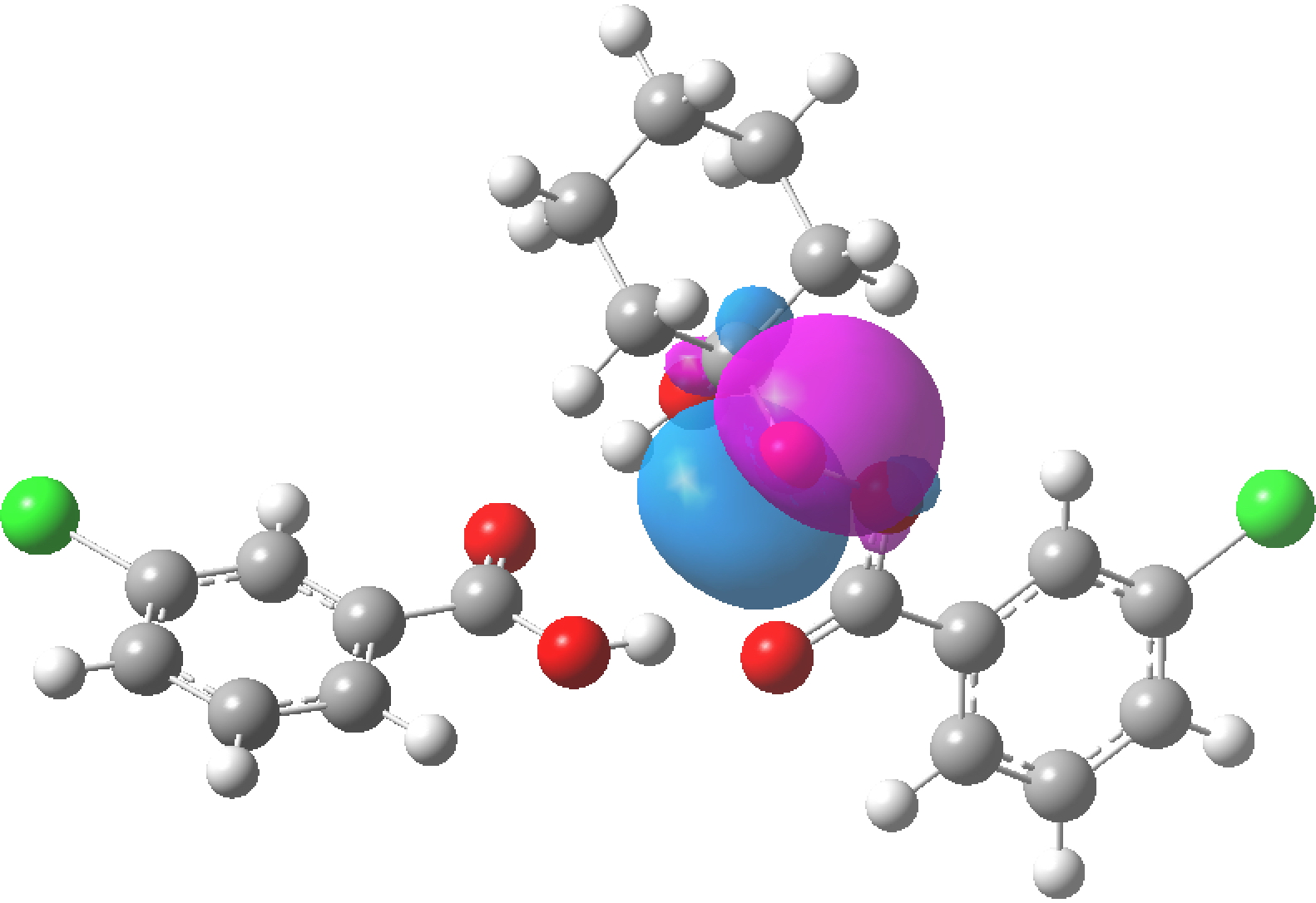
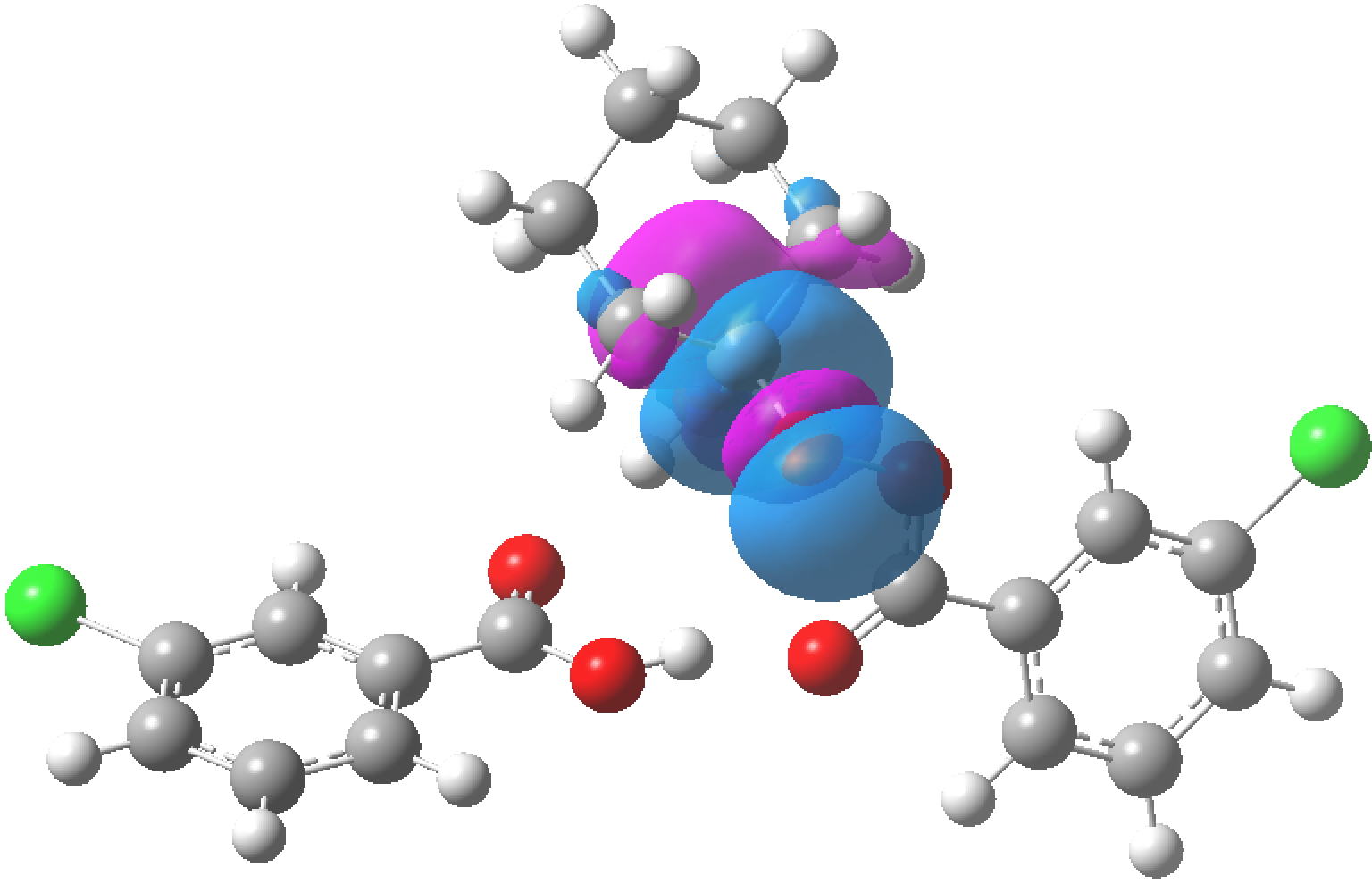
The previous post contained an exploration of the anomeric effect as it occurs at an atom centre X for which the effect is manifest in crystal structures. Here I quantify the effect, by selecting the test molecule MeO-X-OMe, where X is of two types:
- A two-coordinate atom across the series B-O and Al-S, and carrying the appropriate molecular charge such that X carries two lone pairs of electrons (thus the charge is 0 for O, but -3 for B).
- A four-coordinate atom across the series B-O and Al-S, with X-H bonds replacing the lone pairs on this centre in the previous example, and again with appropriate molecule charges (e.g. +2 for SH2).
The donor in the anomeric interaction always originates on the oxygen of the MeO group attached to X. The acceptor is always the X-O σ* empty orbital. The results (table below, ωB97XD/Def2-TZVPP calculation, NBO E(2) in kcal/mol) confirm that as X gets more electronegative, the X-O σ* empty orbital becomes a better acceptor, and so the NBO E(2) interaction energy which quantifies the anomeric interaction gets larger. Eventually (with X=OH2) the donation of electrons into the X-O σ* empty orbital becomes so effective that the X-O bond (in this case O-O) dissociates fully and the NBO perturbation cannot be computed. Also for reference, a “normal” anomeric interaction (such as is found in e.g. sugars) is around 18 kcal/mol. Anything larger than this could be considered especially strong, and anything less than ~10 kcal/mol would be regarded as weak.
| X[cite]10.14469/hpc/1221[/cite]* | |||
|---|---|---|---|
| BH2 | CH2 | NH2 | OH2 |
| 12.5 | 17.7 | 18.5 | dissociates |
| AlH2 | SiH2 | PH2 | SH2 |
| 6.9 | 12.9 | 21.9 | 31.3 |
| B | C | N | O |
| 8.3 | 11.7 | 12.9 | 14.2 |
| Al | Si | P | S |
| 4.8 | 6.6 | 11.2 | 18.2 |
For the entry X=S, the E(2) term is actually larger than for the oxygen. I should note that the Me group itself is not passive in this process. The C-H bonds can also act as significant electron donors, but here I am not going to analyse this additional complexity.
This table reveals that there is nothing special about carbon as an anomeric centre, and here also the normal intimate association with the term anomeric and heterocyclohexanes such as found in sugars.
* Here I introduce a refinement to my normal process of citing the data produced for any specific calculation. Rather than including 16 individual citations for each cell in the table, I have gathered all these calculations into a collection and cite here only the DOI of that collection. When resolved, the individual members of that collection can then be inspected for the actual data.

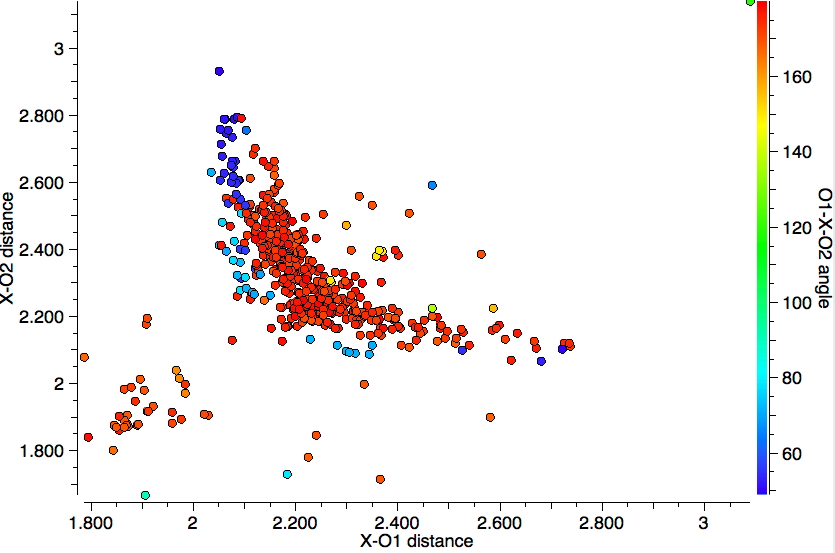
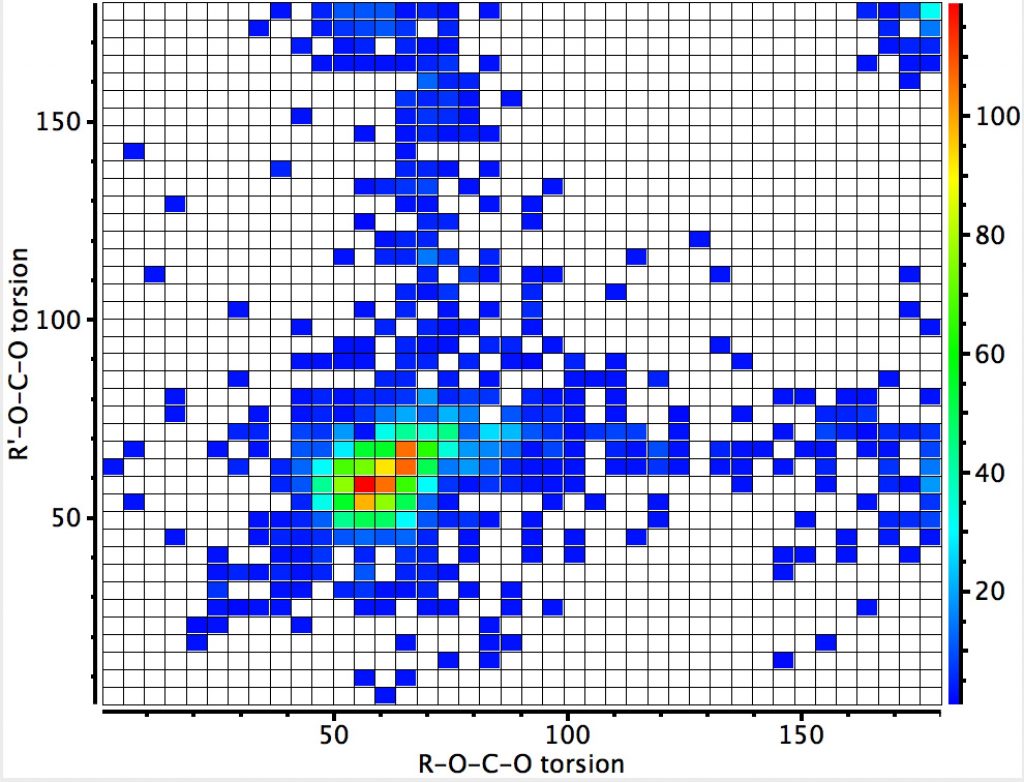
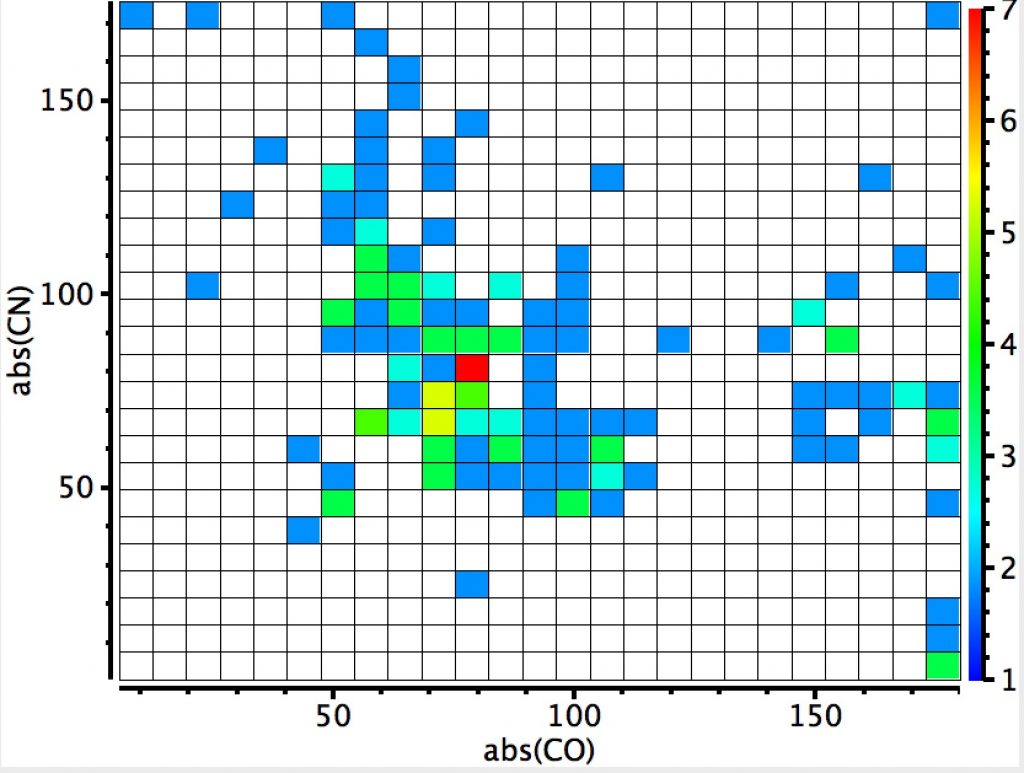 This plot is where both heteroatoms are nitrogen (geminal diamines). There are about twice as many examples, resulting in more distinct clustering. The anomeric hotspot is now around 70° and there are equally populated clusters where only one torsion is ~70°. There is another cluster for which both torsions are 180° (no stereoelectronic alignment of lone pairs) and three small clusters where the torsions are either 180° or 0°. There is finally an intriguing cluster for which both torsions at ~120° (again no stereoelectronics).
This plot is where both heteroatoms are nitrogen (geminal diamines). There are about twice as many examples, resulting in more distinct clustering. The anomeric hotspot is now around 70° and there are equally populated clusters where only one torsion is ~70°. There is another cluster for which both torsions are 180° (no stereoelectronic alignment of lone pairs) and three small clusters where the torsions are either 180° or 0°. There is finally an intriguing cluster for which both torsions at ~120° (again no stereoelectronics). 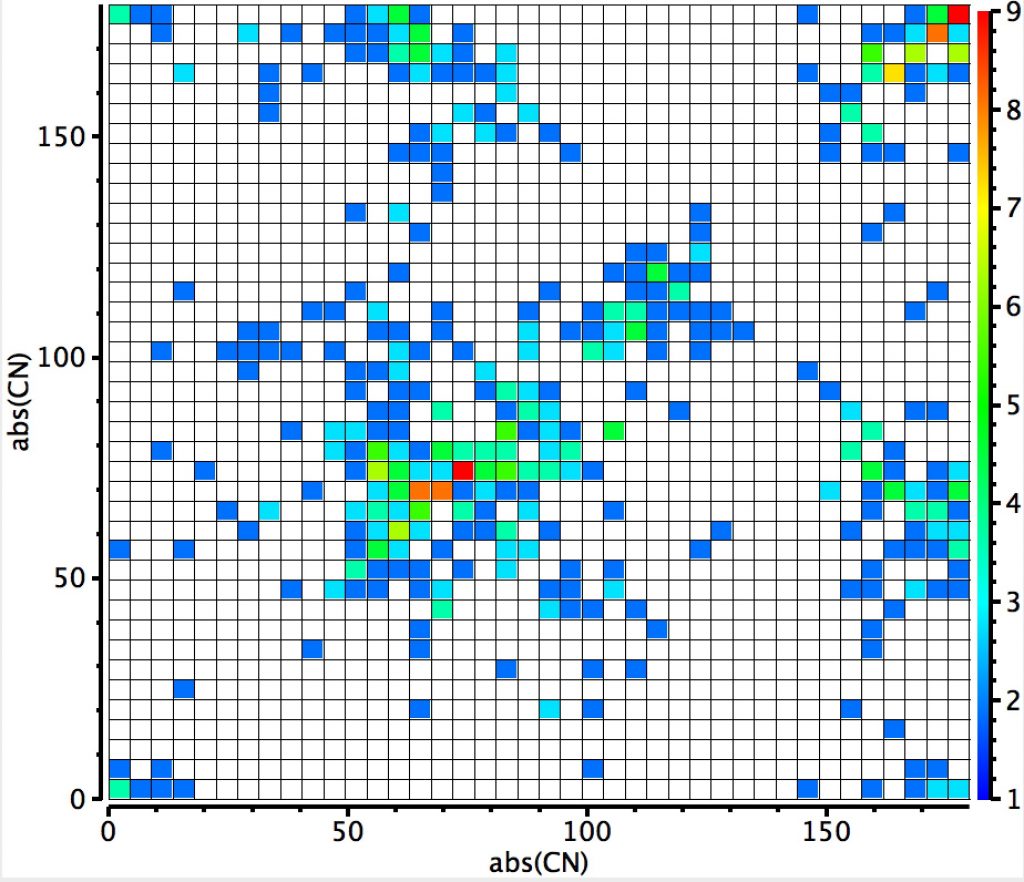
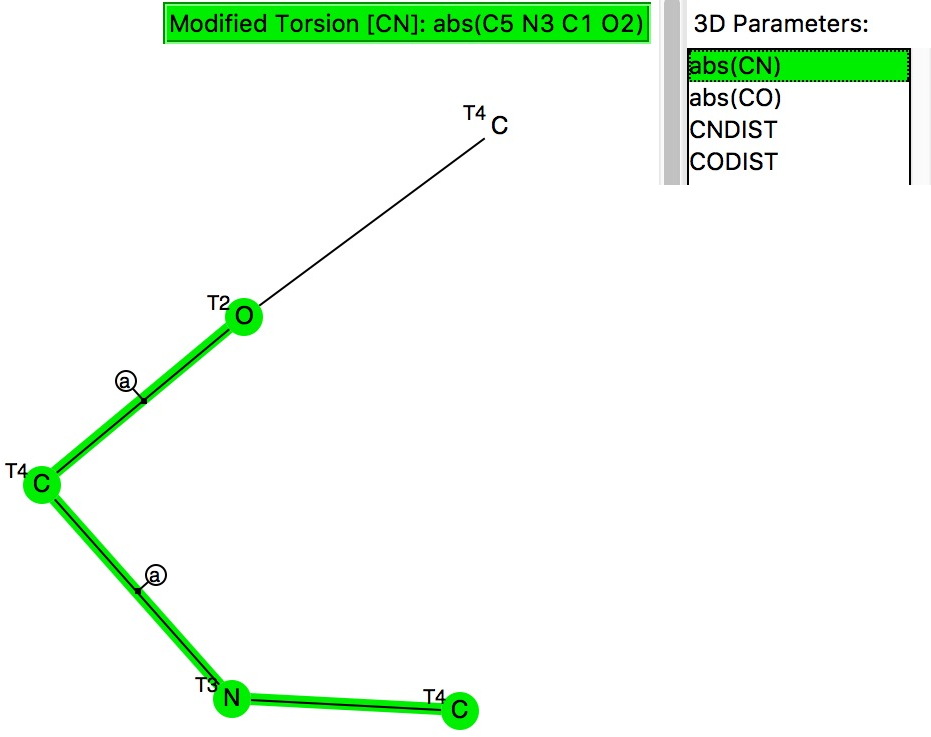
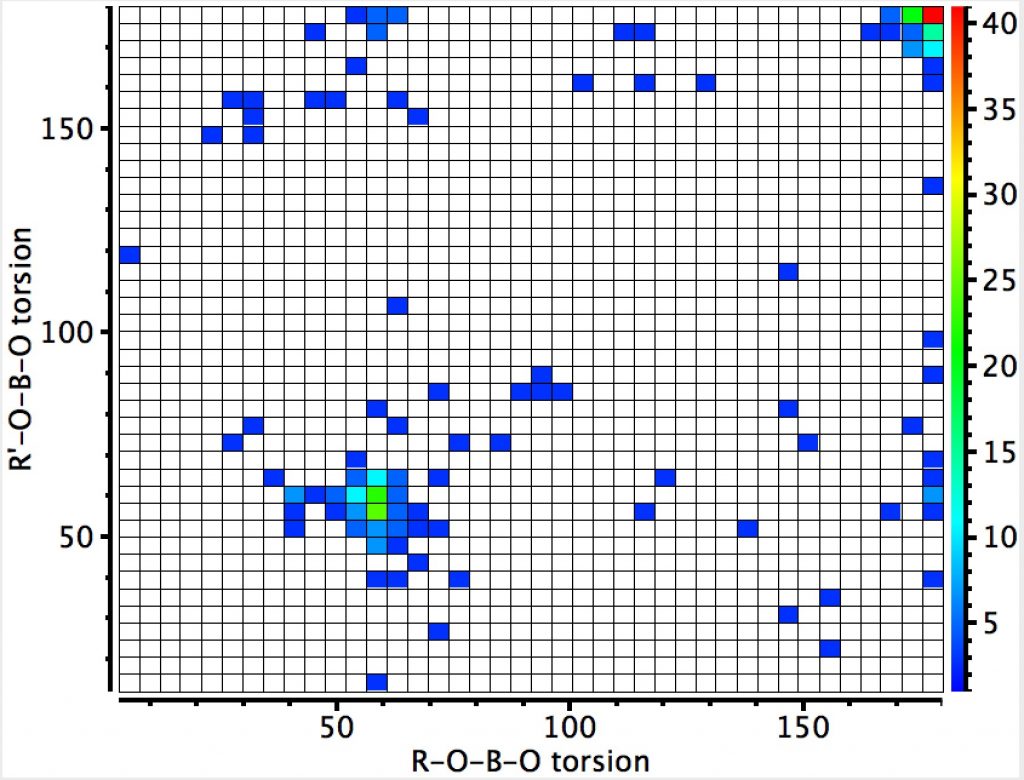 This compares to the carbon-anomeric plot which is shown here for comparison, where the top right cluster of 180° torsions contains proportionately few hits than with boron.
This compares to the carbon-anomeric plot which is shown here for comparison, where the top right cluster of 180° torsions contains proportionately few hits than with boron.
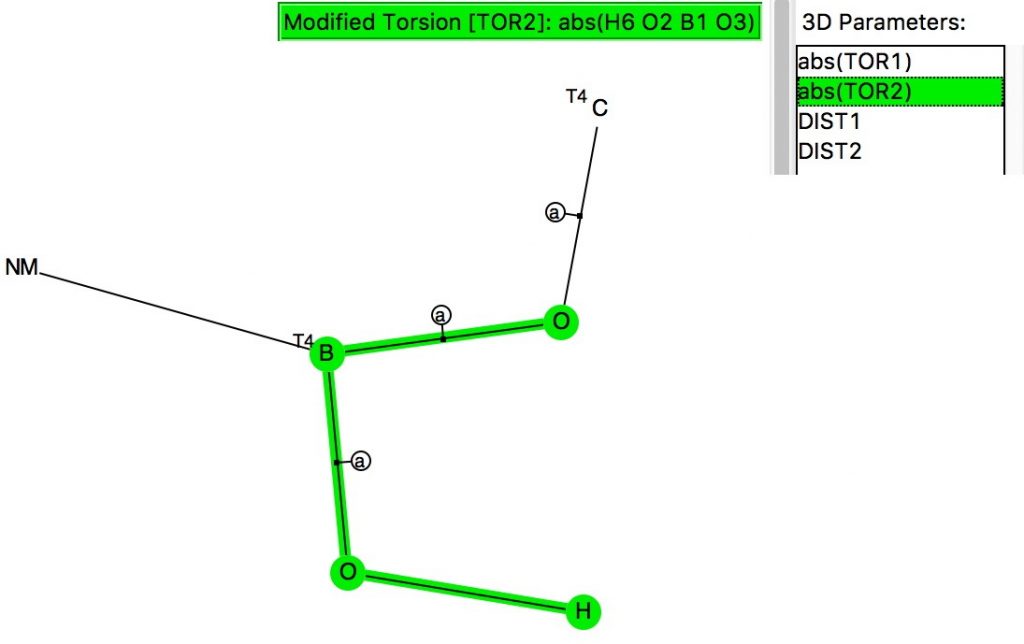
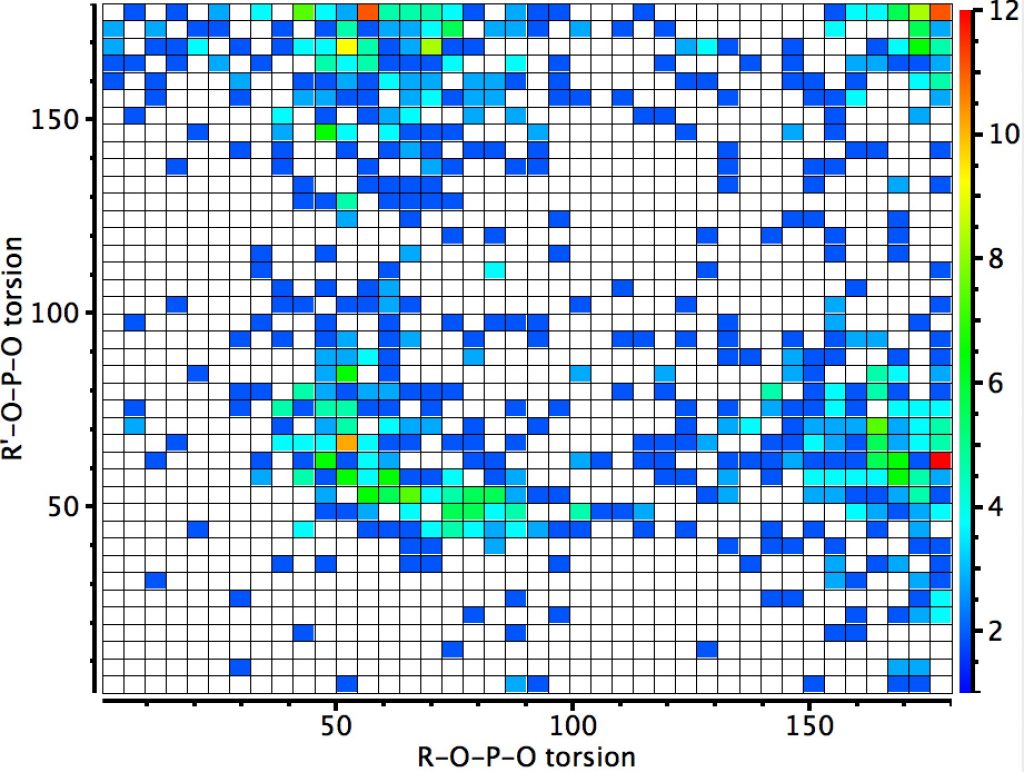
 The next centre is at 4-coordinate silicon. Again three significant clusters are seen; one with two antiperiplanar lone pair alignments with Si-O bonds, and two more with just one such alignment. The previous hotspot for which both measured torsions were 180° is largely absent. So here, the anomeric effect is much stronger. Notice also that whereas the torsions in the region of 60° for the carbon centre lie along a ridge coincident with the diagonal (bottom left to top right), that for the silicon centre show a ridge running orthogonal to the diagonal. An interesting point to follow up perhaps?
The next centre is at 4-coordinate silicon. Again three significant clusters are seen; one with two antiperiplanar lone pair alignments with Si-O bonds, and two more with just one such alignment. The previous hotspot for which both measured torsions were 180° is largely absent. So here, the anomeric effect is much stronger. Notice also that whereas the torsions in the region of 60° for the carbon centre lie along a ridge coincident with the diagonal (bottom left to top right), that for the silicon centre show a ridge running orthogonal to the diagonal. An interesting point to follow up perhaps?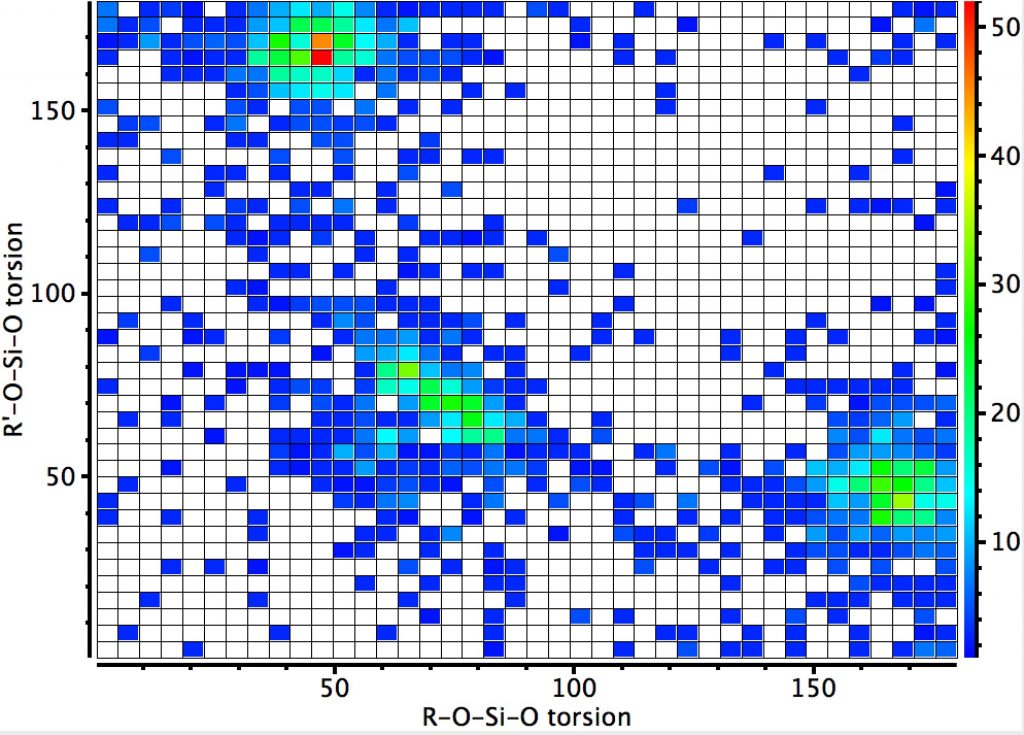
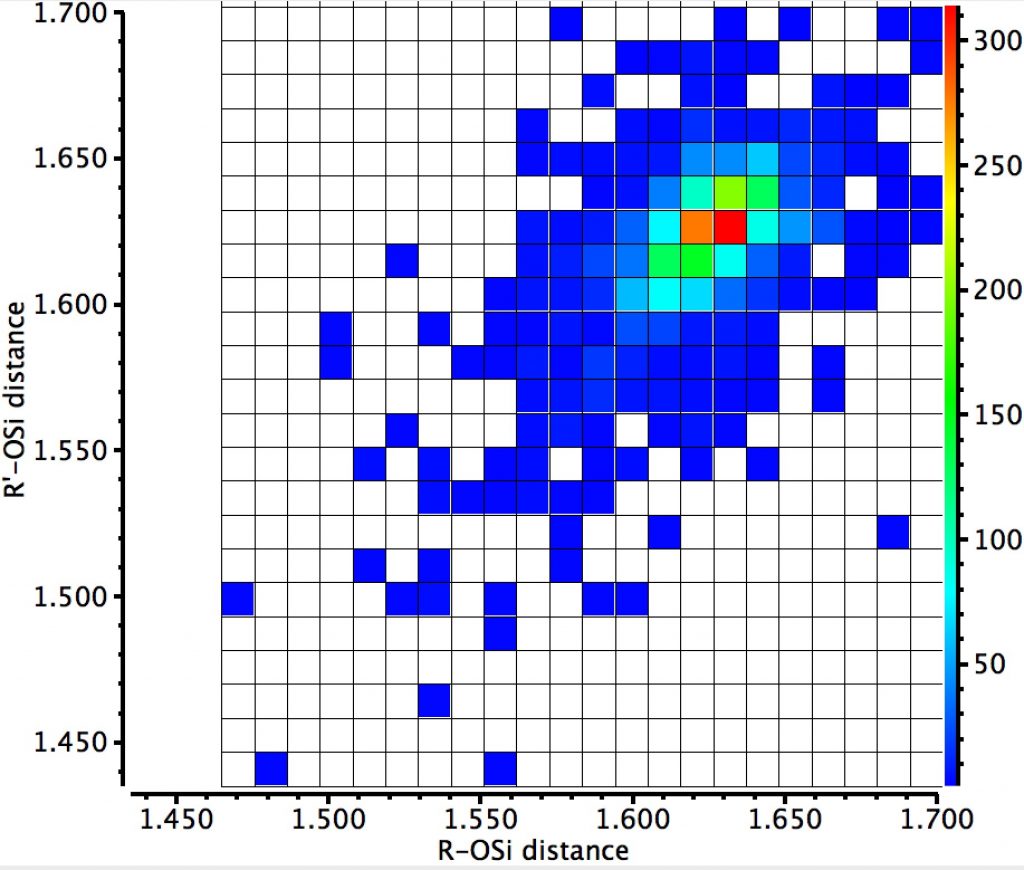
 So this little exploration shows that the anomeric effect, best known for sugars and at a carbon centre, is in fact more general to the adjacent elements.
So this little exploration shows that the anomeric effect, best known for sugars and at a carbon centre, is in fact more general to the adjacent elements.