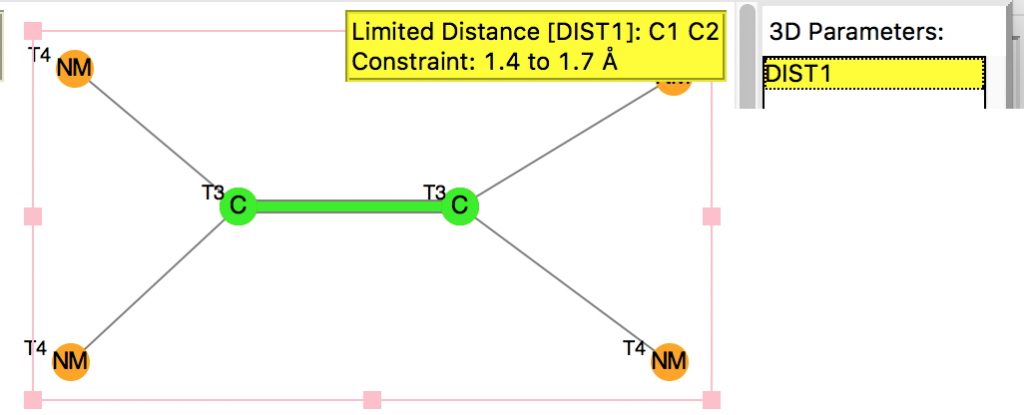
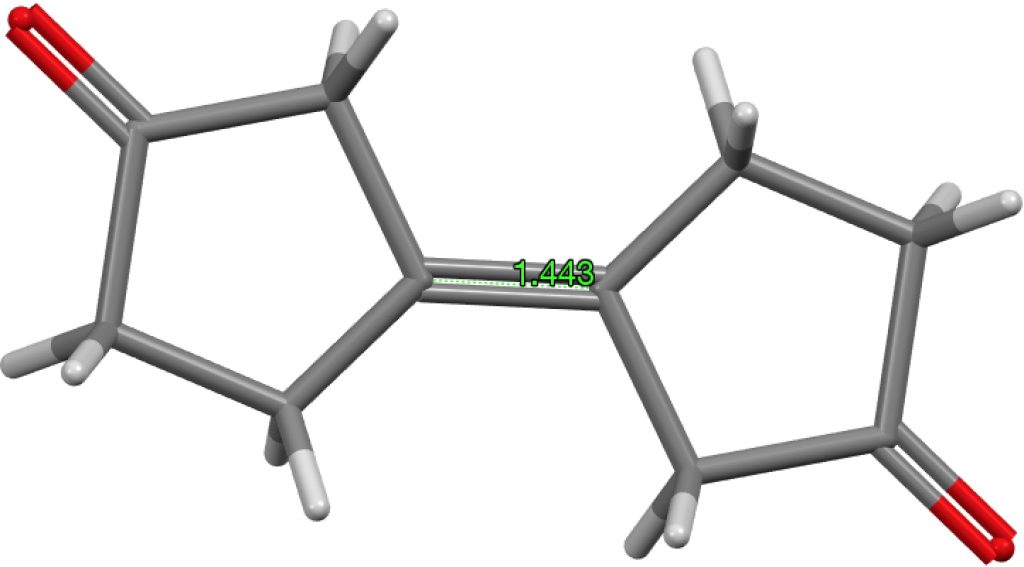
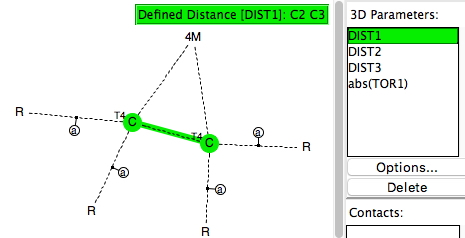
My holiday reading has been Derek Lowe’s excellent Chemistry Book setting out 250 milestones in chemistry, organised by year. An entry for 1920 entitled hydrogen bonding seemed worth exploring in more detail here.
As with many historical concepts, it can often take a few years to coalesce into something we would readily recognise today, and hydrogen bonds are no exception. Wikipedia is another source of the history and it cites a 1912 article as the origin of the term in relation to the solvation of amines[cite]10.1039/CT9120101635[/cite] but also notes that the better known setting of water occurs later in 1920.[cite]10.1021/ja01452a015[/cite] Here I try to capture the essence of the concept with a few diagrams taken from these two articles.
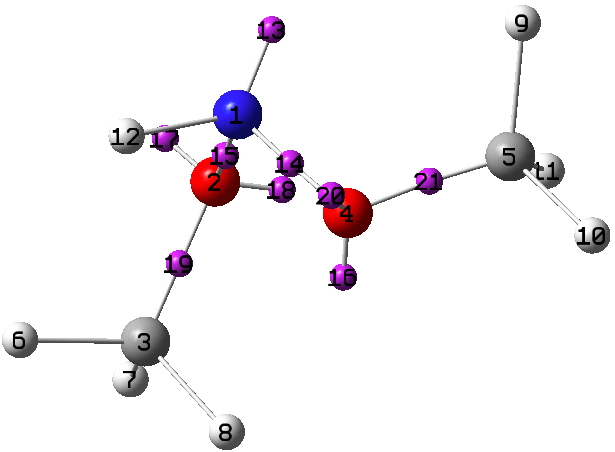
Firstly “The state of amines in aqueous solution“[cite]10.1039/CT9120101635[/cite] which is mostly concerned with the measurement of ionization constants of primary, secondary and tertiary amines. It boils down to the below:
![]()
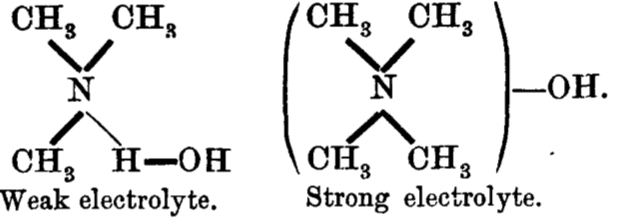
and the connection to ionization is laid out as:

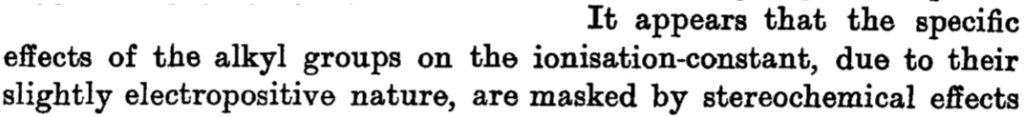
Since in 1912, Lewis’ electron pair theory of the covalent bond had not yet emerged, the authors use the terms “strong union” and “weak union”, and of course it is the “weak union” that we now know of as the hydrogen bond. Some other comments about this seminal diagram:
- The article contains the very explicit and modern term stereochemical, which is used in a manner that suggests it was already common.‡ But there is only a hint at most that the nitrogen atoms might be tetrahedral, or that the “weak union” between (what we now think of as the lone pair on) the nitrogen and the hydrogen of the water is directional.
- The second weak union between the tetramethyl ammonium (which we now describe as a cation) and the hydroxide (now described as an anion; both terms are however implied by the description strong electrolyte) is probably not what we would now call a hydrogen bond, more an intimate ion-pair.
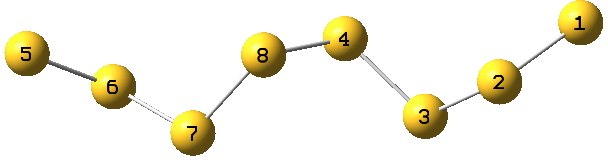
The second article in 1920 on water itself[cite]10.1021/ja01452a015[/cite] is post-Lewis, but perhaps applied in a manner which we would not entirely agree with nowadays. Thus dinitrogen, N≡N is shown as below with just a single connecting bond.
![]()
Then we get the interaction between ammonia and water,† analogous to the example shown above:
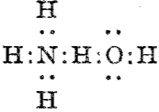
and for water itself:♠
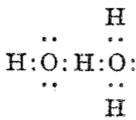
which in each case shows the central hydrogen having what we now call a valence shell of four electrons,♣ and hence more equivalent to the “strong unions” above. This shows that in 1920 chemists were rapidly adopting Lewis’ representations, but not always entirely successfully.
On balance, I think the 1912 article sets out the modern concept of a hydrogen bond representing a weak union to a hydrogen rather better than the Latimer and Rodebush attempt (at least diagrammatically).
‡Stereochemical notation is discussed in this post, and it dates from the 1930s.
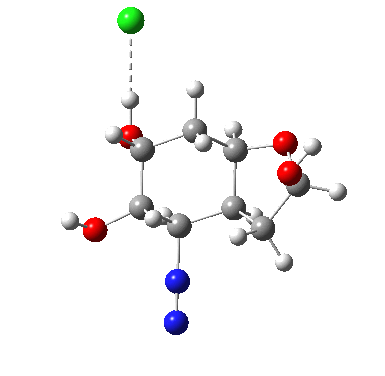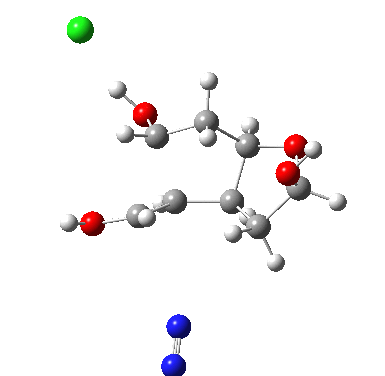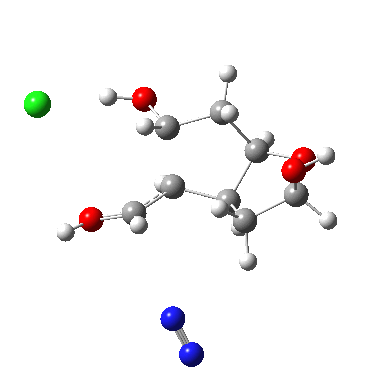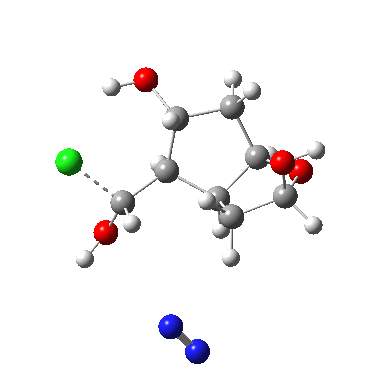
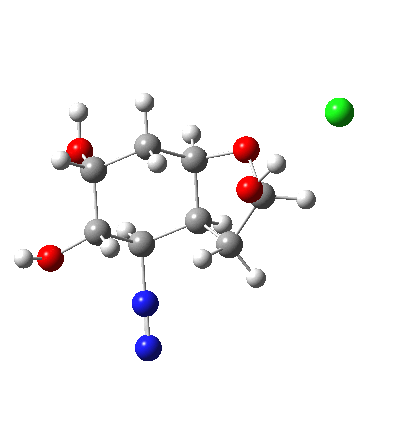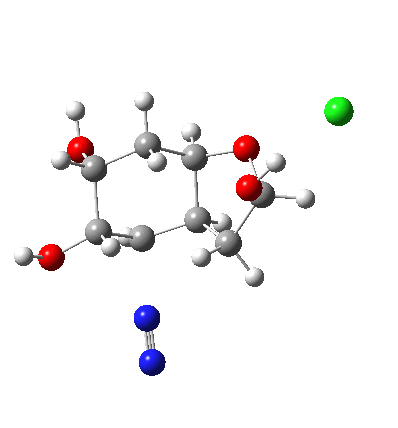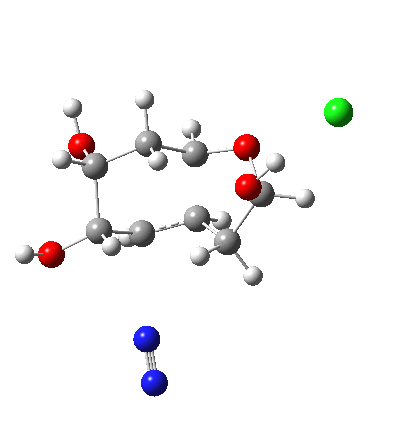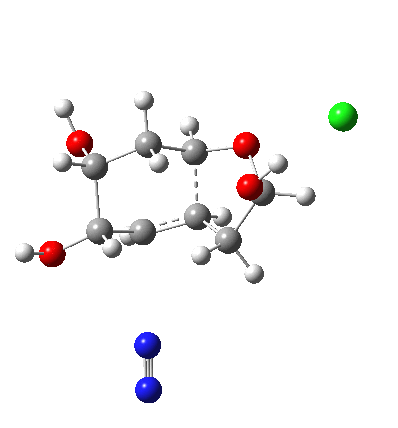
†The modern take is explored here, in which the equilibrium set up between a “weak union” between ammonia and water (the weak electrolyte) and an isomeric “strong union” which represents ionization into an ammonium hydroxide ion-pair (the strong electrolyte) is favoured for the former by ΔG ~6 kcal/mol.
♠The equilibrium between a “weak union” of two water molecules and the fully ionized strong union of hydronium hydroxide favours the former by ΔG ~23 kcal/mol.
♣ This 1920 representation does imply symmetry for the hydrogen, being ~equally disposed between the two oxygens. We now know that such symmetric hydrogen bonding is not unusual (see this post for how to fine-tune a hydrogen bond into this situation) but rather than requiring four electrons as implied in the diagram above, it is now better described as a three-centre-two-electron bond instead.
This post has DOI: 10.14469/hpc/10732