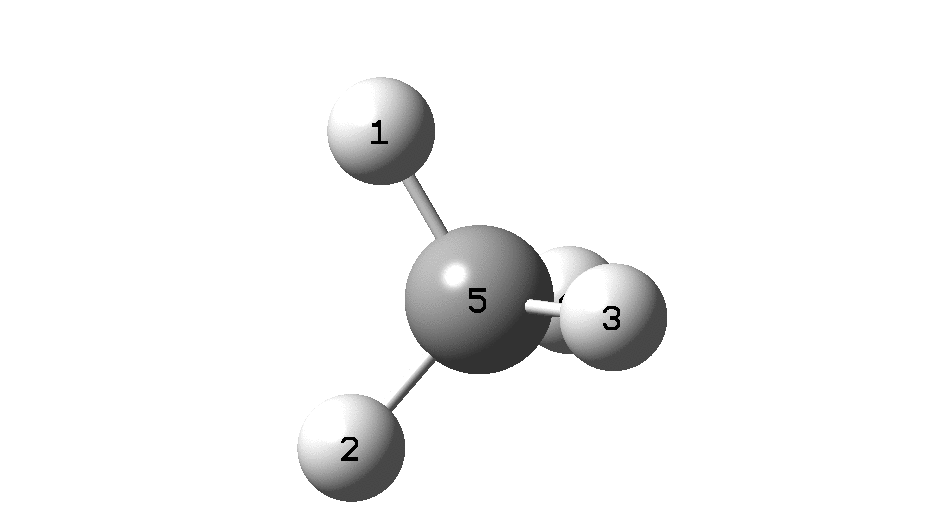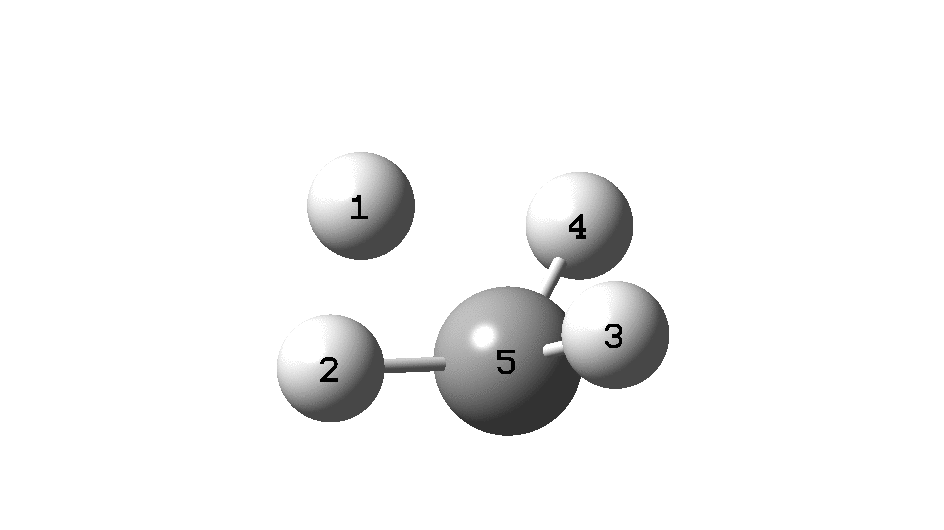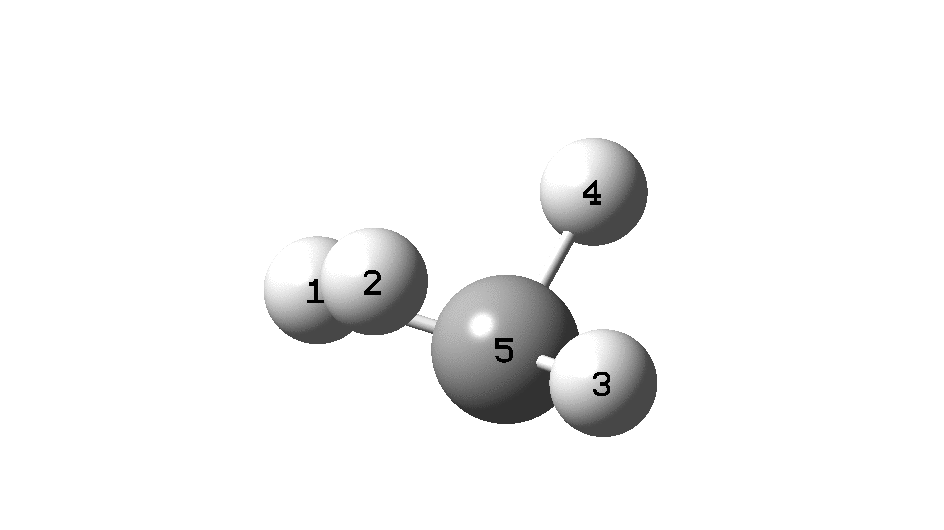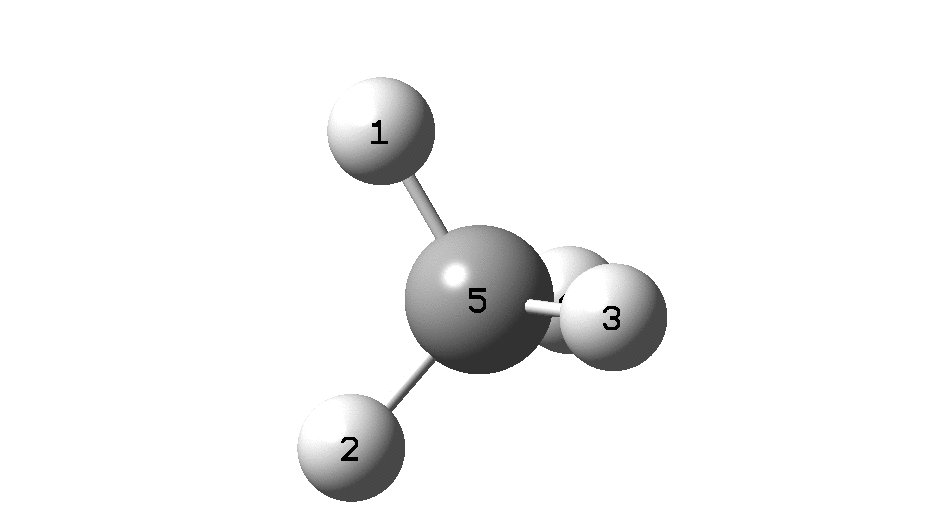The example a few posts back of how methane might invert its configuration by transposing two hydrogen atoms illustrated the reaction mechanism by locating a transition state and following it down in energy using an intrinsic reaction coordinate (IRC). Here I explore an alternative method based instead on computing a molecular dynamics trajectory (MD).
I have used ethane instead of methane, since it is now possible to envisage two outcomes:

An animation of the IRC starting from the located transition state is shown below (DOI: 10.14469/hpc/2331). This is based purely on the computed potential energy surface of the molecule. The IRC is computed from the forces experienced on the atoms as they are displaced from an initial set of coordinates corresponding to the located transition state and then following the direction indicated by the eigenvectors of the negative force constant required of a transition state. Importantly, there is no time component; the path is based entirely on energies and forces.
Next, a molecular dynamics simulation (ωB97XD/6-31G(d,p), DOI: 10.14469/hpc/2330). This uses the ADMP method, which requests a classical trajectory calculation using the “atom-centered density matrix propagation molecular dynamics model”. This integrates kinetic energy contributions from the molecular vibrations and so explicitly now includes a time component. In this example the evolution of the system from the transition state is charted over a period of 100 femtoseconds (1000 integrated steps). As it happens this is a relatively short period of evolution; sometimes periods of picoseconds may be required to get a realistic model, especially if one is also dealing with explicit solvent molecules (of which perhaps 500 might be required).
Such explicit inclusion of the kinetic energy from molecular vibrations in effect allows the molecule to “jump” over shallow barriers. In this case, the barrier for a [1,2] hydrogen shift from the methyl group to the adjacent carbene (watch atom 8). Simultaneously, the path taken by two hydrogens no longer corresponds to their transposition but to their elimination as a hydrogen molecule. So this quite different outcome from the IRC is very probably also a much more realistic one.
If the MD method is so much more realistic than the IRC, then why is it not always used? The simple answer is computational time! For this very small molecule and using quite a modest basis set (6-31G(d,p)), the relatively short 1000 time steps took about three times as long to compute as the IRC. The factor gets worse as the size of the molecule increases and the number of time steps for a realistic result increases. Perhaps, as technology gets better and new computer architectures emerge, MD simulations of ever increasingly complex reactions will become far more common. In ten years time, I expect most of the examples on this blog will use this method!