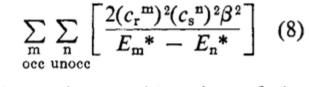The previous posts produced discussion about the dipole moments of highly polar molecules. Here to produce some reference points for further discussion I look at the dipole moment of glycine, the classic zwitterion (an internal ion-pair).
Dielectricrelaxation studies of glycine–water mixtures yield values that range from 15.7D[cite]10.1039/TF9635900085[/cite] to 11.9D[cite]10.1016/j.molliq.2004.08.001[/cite] although these have to be derived using various approximations and assumptions for up to 4 independent Debye processes. Before proceeding to calculations, I looked at the properties of ionized amino acids in the solid state, using the following search query for the Cambridge structure database (CSD).
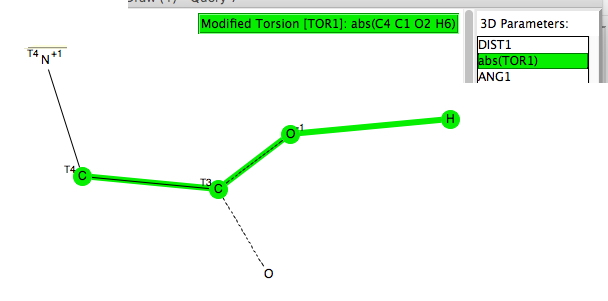
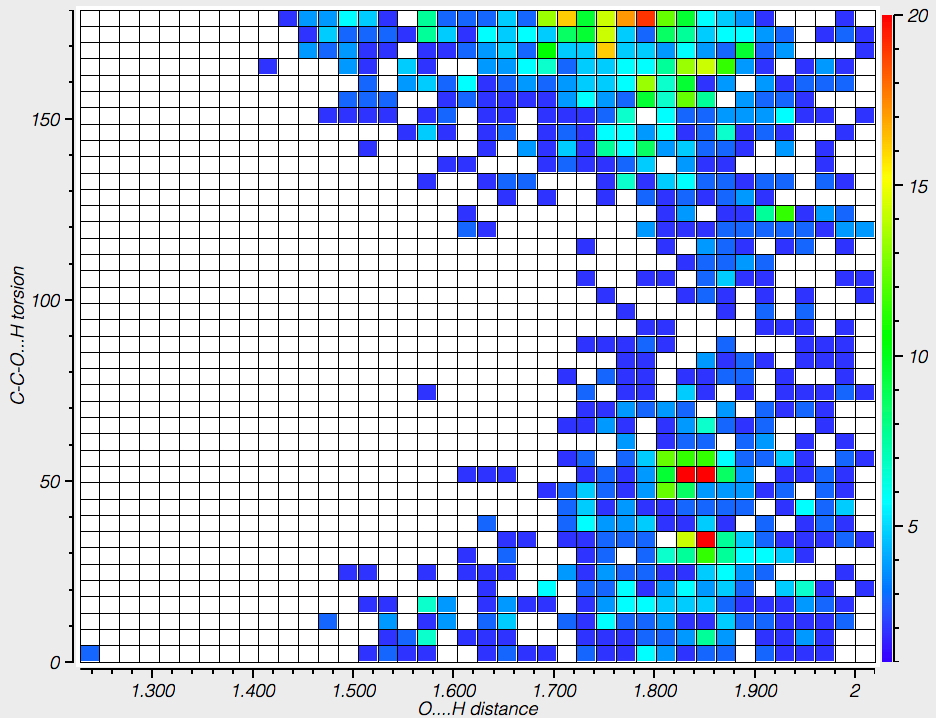
The distance measures hydrogen bonds to the carboxylate oxygens and the torsion their orientation. The O…H hydrogen bond distances vary between 1.7-1.85Å, which are short. The orientation of the hydrogen bond can be to the in-plane oxygen “σ-lone pair” (torsion 0 or 180°) and also an out-of-plane ~π form (torsion ~60-90°).
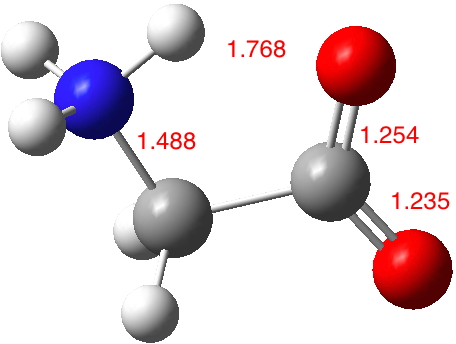
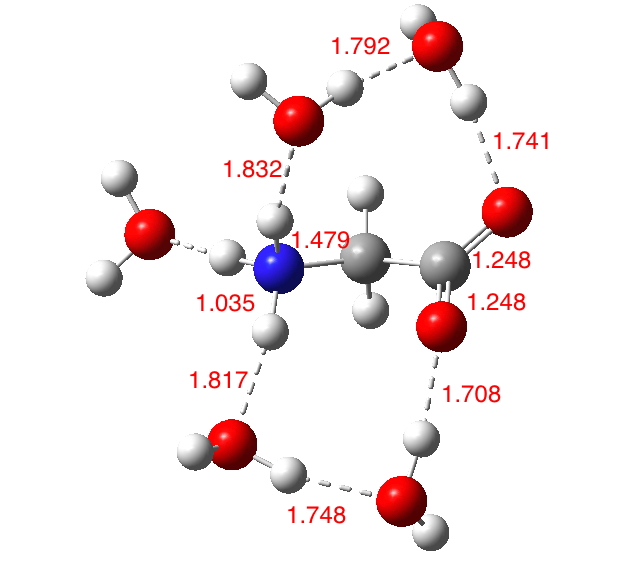
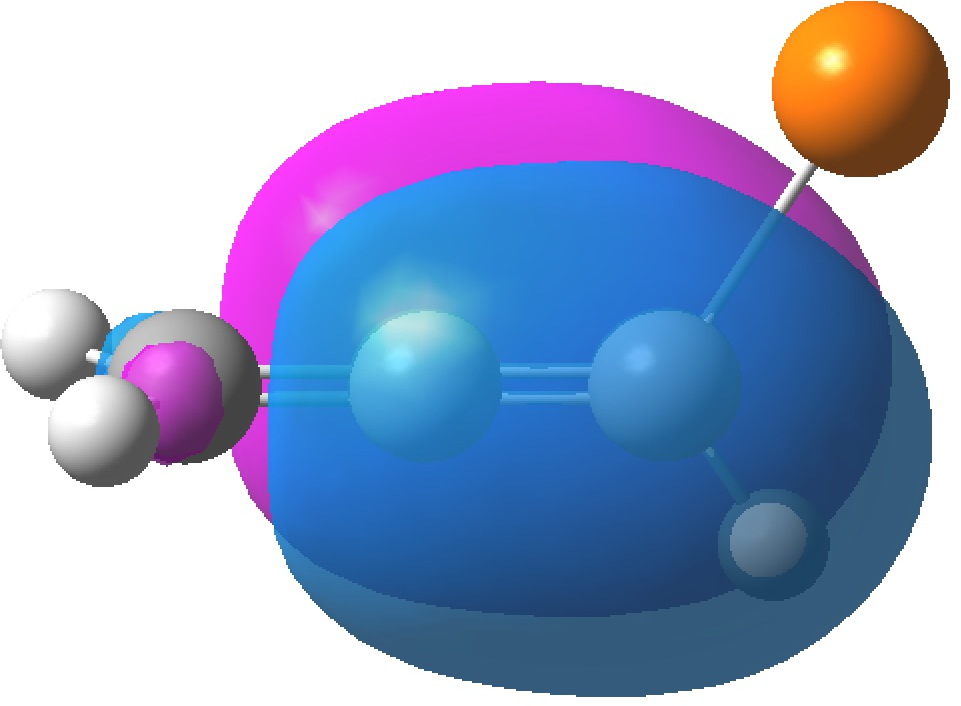
In aqueous solution, it is normally assumed that glycine sustains five such strong H-bonds (three to the H3N+ group and two[cite]10.1021/jp020957j[/cite] to the carboxylate anion), forming a polarised “salt bridge” across the ion-pair. Two model types were subjected to calculation using ωB97XD/Def2-TZVPP/SCRF=water. Aqueous glycine without any added explicit water molecules yields a dipole moment of 12.9D (DOI: 10.14469/hpc/2000), which is within the range noted above.‡
The solvated form is shown below, in one specific conformation of the three studied (ωB97XD/Def2-TZVPP/SCRF=water). The calculated O…H hydrogen bond lengths fall into the range revealed from crystal structures. The calculated dipole moments range from 12.6 (DOI: 10.14469/hpc/2007), 15.3 (DOI: 10.14469/hpc/2006) and 14.9D (DOI: 10.14469/hpc/2005), which is a modest increase over the model with no explicit water molecules. The actual dipole is of course a Boltzmann average over these and other as yet unexplored conformations, as well as other values for the number of water molecules.
Given the difficulties in interpreting the dipole moment of a complex Debye system such as hydrated glycine, the agreement between the limited range of solvated models and the measured values seems reasonable, and provides at least some measure of “calibration” for the polar molecules commented on previously.
‡Optimized with the solvent field on. If a vacuum model is used, the proton transfers from the N to the O.
I am completing my survey of the vote for molecule of the year candidates, which this year seems focused on chemical records of one type or another.
The first article[cite]10.1002/chem.201601916[/cite] reports striving towards creating a molecule covering a complete column of the period table. In this case, group 7, containing N, P, As, Sb, Bi and Mc. Only the first four of these were incorporated, although the prospects of extending this to five seem good (and to six extremely unlikely). The structure of this pnictogen chain is referenced here: DOI: 10.5517/CCDC.CSD.CC1LHPJ9 and I have demurred from a calculation.
The second article[cite]10.1021/jacs.6b02590[/cite] relates to what might be called hypercoordination, and the achievement of what is felt is a maximum value of 16 to a single metal. I thought I might approach this one by searching the Cambridge structure database (CSD) by specifying any metal with a coordination number 16 as the search query. However, I was foiled in this query because the search software (Conquest) allows a maximum value of only 15! So instead I list the total number of hits retrieved for coordination numbers of 10-15: 25224, 4753, 8856, 2492, 839, 348 respectively.
These totals have to be taken with some caution; the coordination number of what may often be very weak interactions may be often determined by human chemical perception rather than hard and fast rules. Nevertheless, the assignment of 348 molecules to having a coordination number of 15 is still a remarkably high number. If I can persuade CCDC to allow searches with 16, who knows what other candidates might emerge to rival this one, DOI: CCDC.CSD.CC1KFCQ2
The final candidate[cite]10.1039/C6SC01726F[/cite] is the only one where no measured coordinates are reported, with the title “Preparation of an ion with the highest calculated proton affinity: ortho-diethynylbenzene dianion”. There high level theoretical and computational modelling is reported to which I cannot add anything useful.
The common theme emerging of my review is that most of the candidates have crystal structures to which I have been able to occasionally add some computed quantum mechanical properties to try to tease out some other aspects of their character. It is also nice to be able to cite a persistent identifier (DOI) that leads directly to the 3D coordinates for the structures. My first ever post to this blog in 2008 addressed one solution on how such immediacy might be achieved and it is nice to see this now as a mainstream aspect of chemical publishing.
Following on from a search for long C-C bonds, here is the same repeated for C=C double bonds.
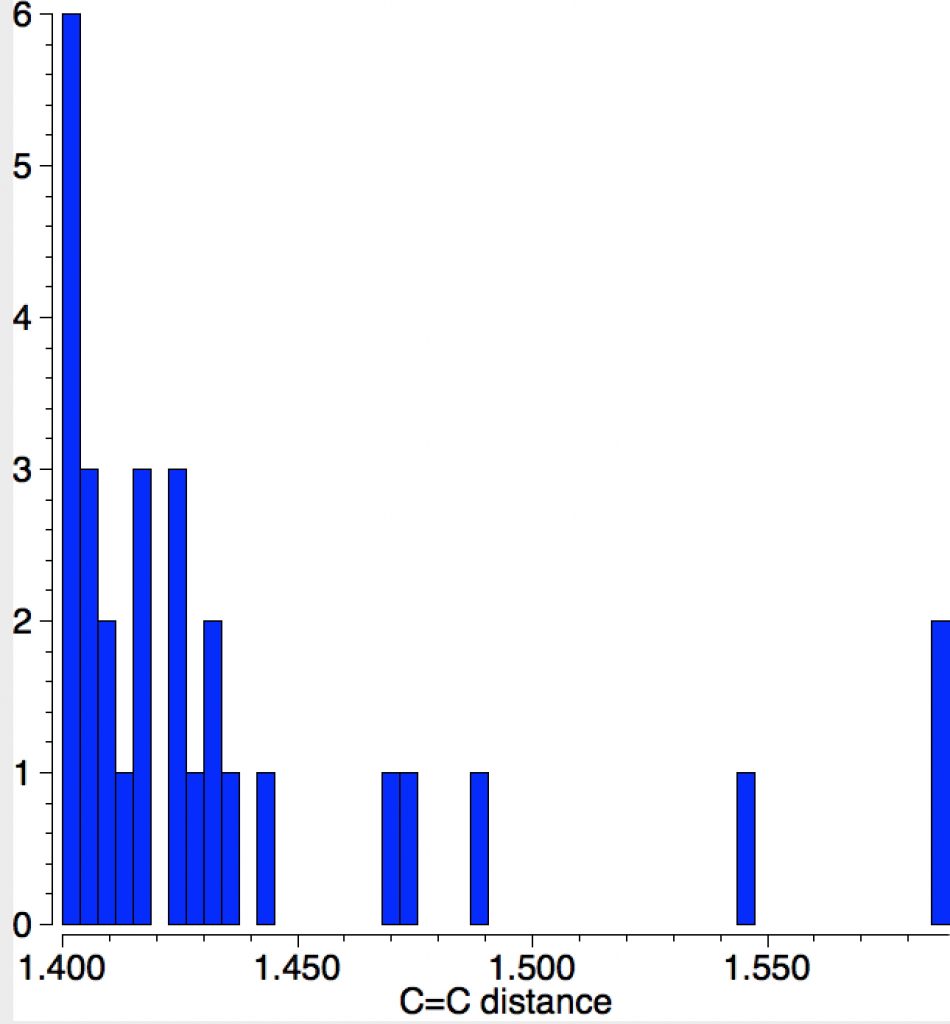
The query restricts the search to each carbon having just two non-metallic substituents. To avoid conjugation with these, they each are 4-coordinated; the carbons themselves are three-coordinated. Further constraints are the usual no disorder, no errors and R < 0.1 and the C=C distance > 1.4Å (the standard value is ~1.32-1.34Å). The search query is deposited as DOI: 10.14469/hpc/1959[cite]10.14469/hpc/1959[/cite]
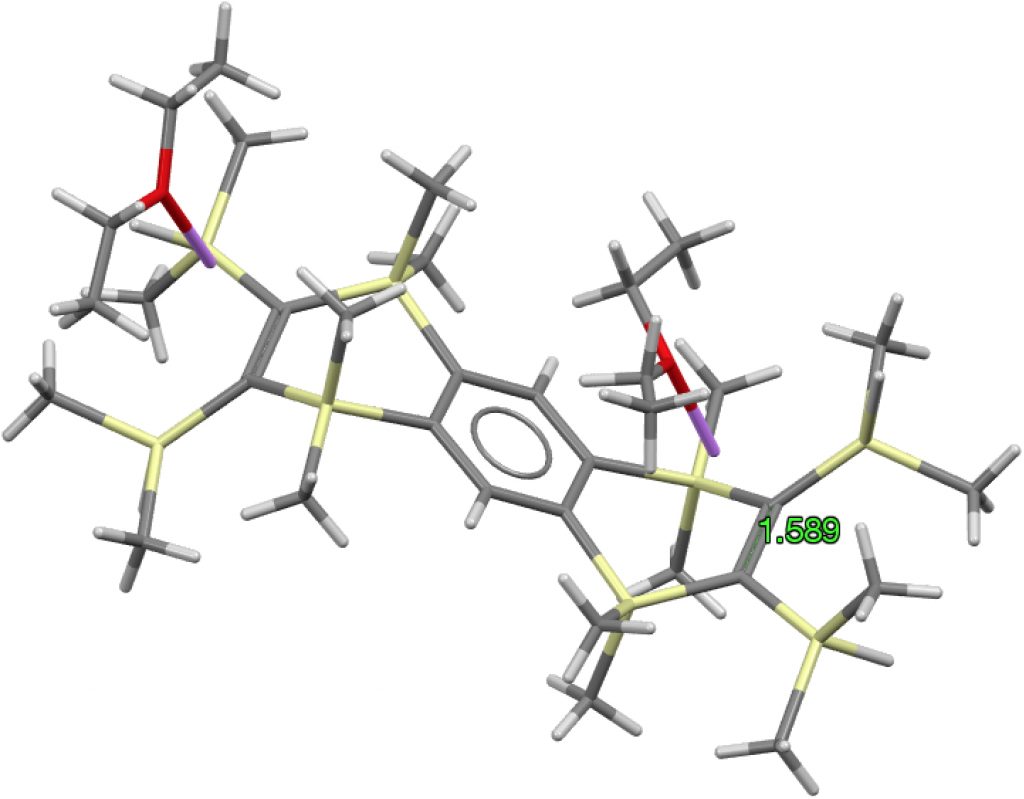
The apparent longest example is LIRVEN, DOI: 10.5517/CC4R2MK[cite]10.5517/CC4R2MK[/cite] with a value of 1.589Å, longer than most C-C single bonds! Closer inspection reveals the presence of lithium cations, and so the molecule bearing the C=C bond must sustain two negative charges. So this apparent C=C bond is in fact anionic, with one electron going into each of the π* orbitals, thus lengthening the CC bond.‡ Not a true example of a neutral C=C bond[cite]10.1016/S1387-7003(99)00136-7[/cite] but it now becomes interesting for what its spin state might be. Is it a biradical or a triplet for example? One to be investigated further I fancy! Another example of this type is QUKCEE[cite]10.1246/bcsj.73.1461[/cite]
This next FAZWIM has a C=C length of 1.546Å. It is an old structure (1986), and comes without attached hydrogen atoms. Although drawn with no hydrogens on the central C=C bond, the length suggests this molecule is simply mis-assigned.†
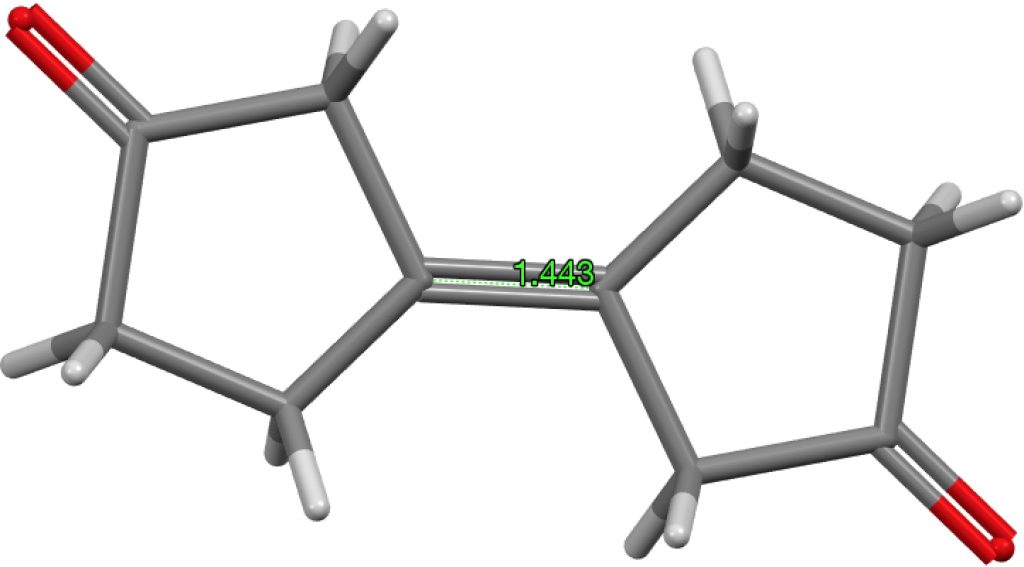
The final example I will highlight is pretty ordinary looking and published in 2016 as a private communication; ALOVOO, DOI: 10.5517/CCDC.CSD.CC1LJSWS[cite]10.5517/CCDC.CSD.CC1LJSWS[/cite] with a C=C length of 1.443Å. Again no obvious reason for the bond to be longer than normal.‡†
In hunting for such unusual deviations from the norm, the most obvious explanation is normally some anomaly in the crystallographic analysis. Although the CSD (crystal structure database) is a very heavily curated resource, it seems unlikely that each deposition would be carefully inspected for its chemistry, and this must be our task here. But such anomalies can themselves point to interesting or unusual chemistry, which in turn can be subjected to quantum computation to see if either the unusual value can be replicated or other reasons identified. In this case, this exercise can been conducted by a human, but one can easily envisage the entire process being automated on a far larger scale. The future?
‡ In fact the stoichiometry shows each “double bond” is actually a di-anion, with two electrons entering each of the the π* orbitals.
†A calculation on the singlet state for the structure as drawn (ωB97XD/Def2-TZVPP, DOI: 10.14469/hpc/1960) gives a bond length of 1.342Å, i.e. that expected for a double bond. The triplet state is similar in energy, but with a much longer central bond length of 1.476Å, DOI: 10.14469/hpc/1962 but the geometry at the carbons is planar and not bent as shown above. The quintet state is 1.45Å and is again planar, doi 10.14469/hpc/1963. So calculations on FAZWIM strongly suggest the structure as shown is an error.
‡†The computed value is 1.324Å, perfectly normal. DOI: 10.14469/hpc/1966[cite]10.14469/hpc/1966[/cite]
Bromoallene is a pretty simple molecule, with two non-equivalent double bonds. How might it react with an electrophile, say dimethyldioxirane (DMDO) to form an epoxide?[cite]10.1039/C6CC06395K[/cite] Here I explore the difference between two different and very simple approaches to predicting its reactivity.
Both approaches rely on the properties of the reactant and use two types of molecule orbitals derived from its electronic wavefunction. The first of these is very well-known as the molecular orbital (MO), which has the property that it tends to delocalise over all the contributing atoms (the “molecule”). MOs are often used in this context; the highest energy occupied MO is thought of as being associated with the most nucleophilic (electron donating) regions of the molecule and so such a HOMO would be expected to predict the region of nucleophilic attack. The second is known as the natural bond orbital (NBO), which is evaluated in a manner that tends to localise it on bonds (the functional groups or reaction centres) and atom centres. What do these respective orbitals reveal for bromoallene?
The MOs
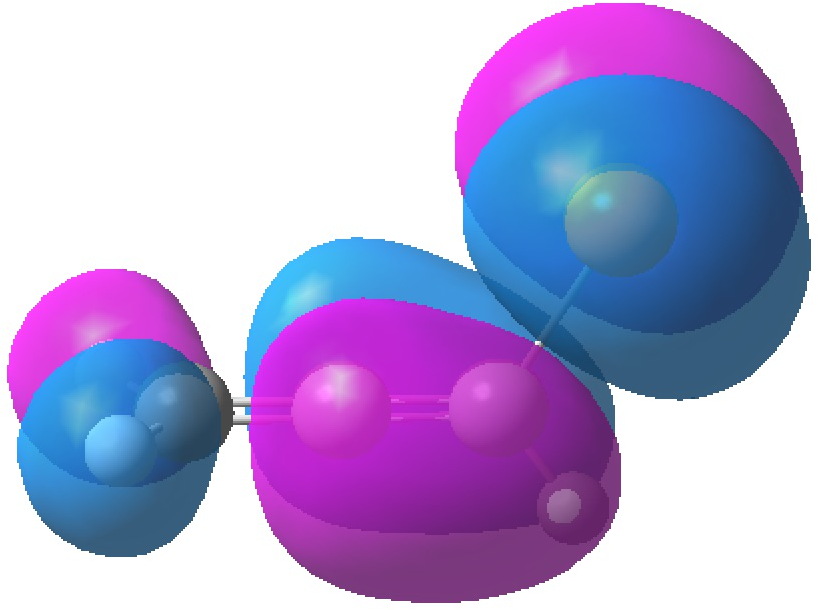
HOMO, -0.3380
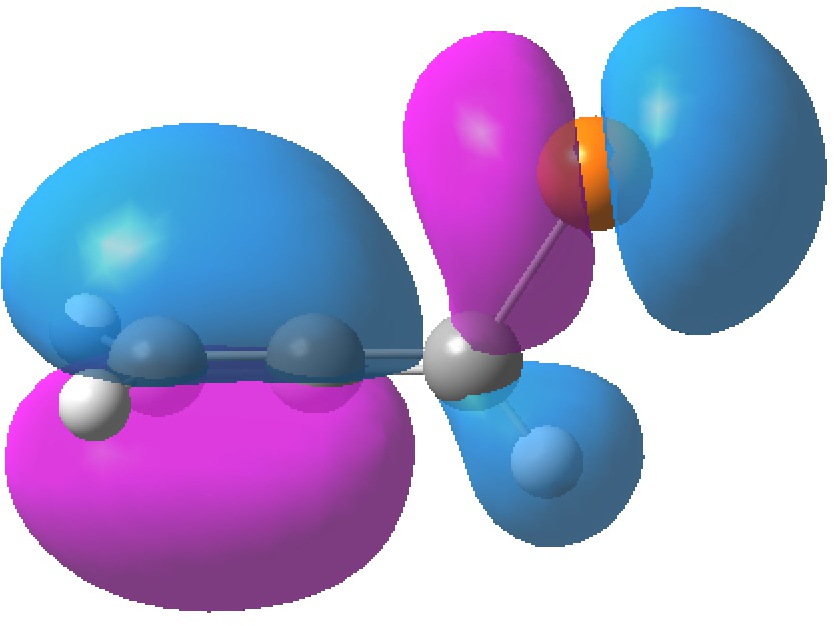
HOMO-1, -0.3692 au
Click for 3D
Click for 3D
The NBOs
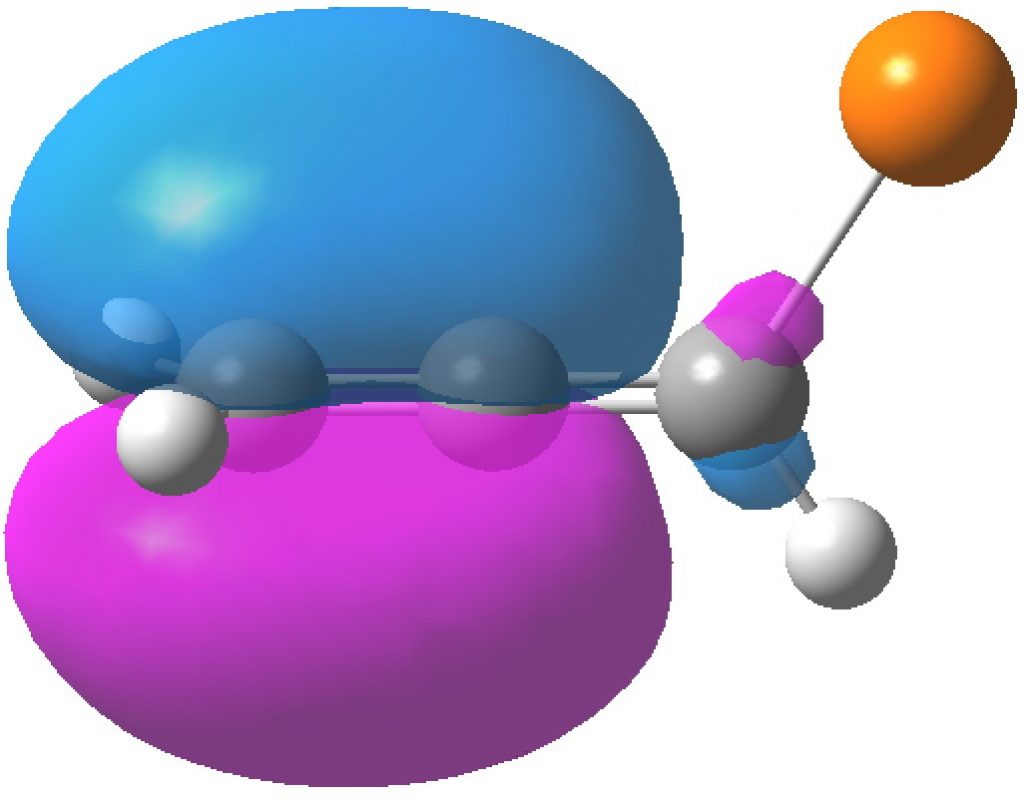
HONBO, -0.3769
HONBO-2, -0.3898
Click for 3D
Click for 3D
The table above shows the energies (in Hartrees) of the four relevant orbitals. The less negative (less stable) the orbital, the more nucleophilic it is. The (heavily) delocalized HOMO is located on the C=C bond bond carrying the C-Br bond, Δ1,2 alkene, but it also has a large component on the Br. The more stable HOMO-1 is located on the C=C bond located away from the Br, the Δ2,3 alkene and also with a (different type of) component on the Br.
In contrast, the HONBO is located on the Δ2,3 alkene and it is the HONBO-2 that is on the Δ1,2 alkene. Both these orbitals have very little “leakage” onto other atoms, they are almost completely localised.
Well, now we have a problem since these two analyses lead to diametrically opposing predictions! So what does experiment say? A recent article[cite]10.1039/C6CC06395K[/cite] addresses this issue by isolating the initially formed epoxide from reaction with DMDO and characterising it using crystallography. But here comes the catch; such isolation only proved possible if the allene was also substituted with large sterically bulky groups such as t-butyl or adamantyl. And the isolated product was the Δ1,2 epoxide. So does that mean that the MO method was correct and the NBO method wrong? Well, not necessarily. Those large groups play an additional role via steric effects. To factor in such effects one has to look at the transition state model for the reaction rather than depending purely on the reactant properties. And the steric effects in this case appear to win out over the electronic ones.[cite]10.1039/C6CC06395K[/cite]
The Klopman[cite]10.1021/ja01004a002[/cite]-Salem[cite]10.1021/ja01005a001[/cite] equation (shown in very simplified, and original, form below for just the covalent term) casts some light on what is going on. This term is a double summation over occupied/unoccupied (donor-acceptor) orbital interactions, involving the coefficients of the orbitals (the overlap integrals in effect) in the numerator and the energy difference between the occupied/unoccupied orbital pair as denominator.
Performing such a double summation is rarely attempted; instead the equation is reduced to just one single term involving the donor of highest energy and the acceptor of lowest energy, ensuring the energy difference is a minimum and hence the term itself is (potentially) the largest in the summation. There is still the issue of the orbital coefficients, and here we get to the crux of the difference between the use of MOs and NBOs. You can see by inspection that the two π-MOs for bromoallene have different coefficients on the two atoms of interest, the two carbons of the double bond. One really has to evaluate the size of this term in the summation by using quantitative values for the respective coefficients and to very probably include the further terms in the summation for any other orbitals which also have significantly non-zero coefficients on these two atoms. But with the NBOs, the localisation procedure used to derive them has reduced the coefficients to just the carbon atoms and effectively no other atoms; all the other terms in the double summation in effect do drop out entirely. So with NBOs, the only number that matters is the energy difference between the occupied/empty orbitals (the denominator). But since the acceptor (the electrophile, DMDO in this case) is the same for both regiochemistries, things reduce even further to just comparing the donor energies for the two alternatives (Table above). The higher/less stable of these will have the greater contribution in the Klopman-Salem equation.
This little molecule teaches the important lesson that electronic and steric effects both play a role in directing reactions, and in this system they may well oppose each other. Simple interpretations based on reactant orbitals may give only a partial and even potentially misleading answer.