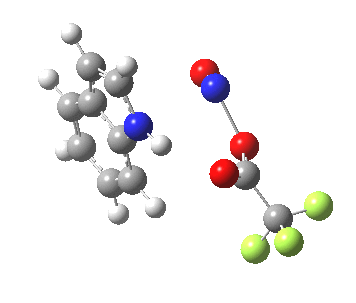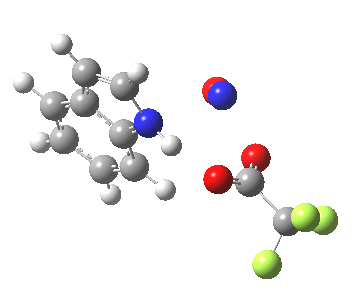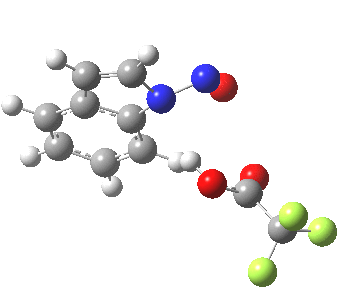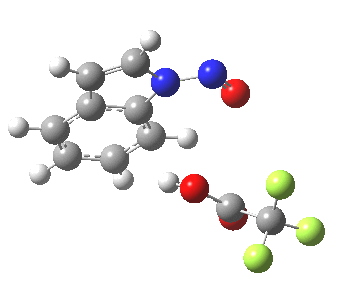
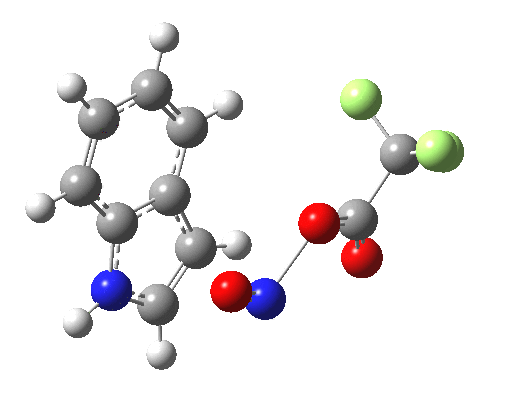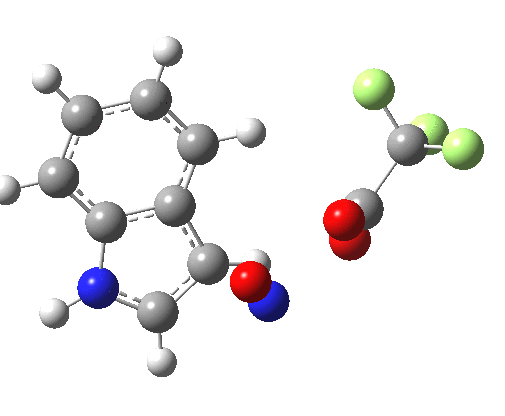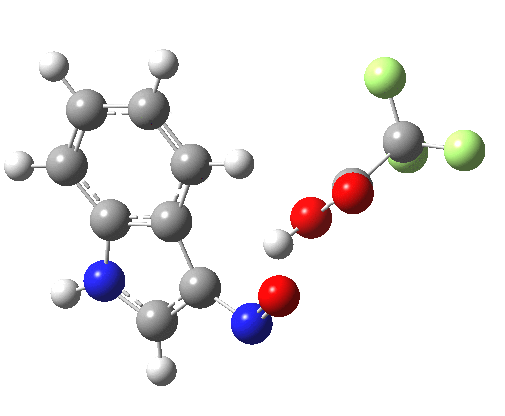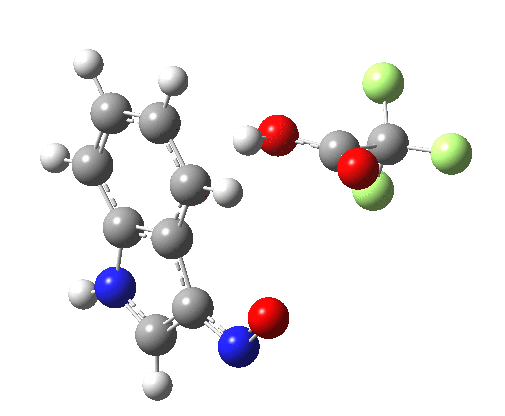
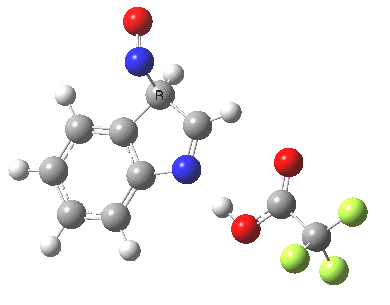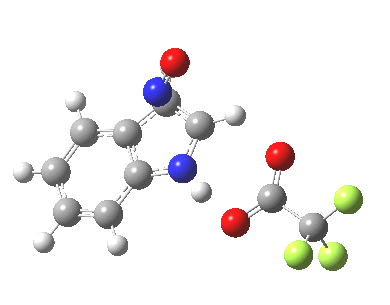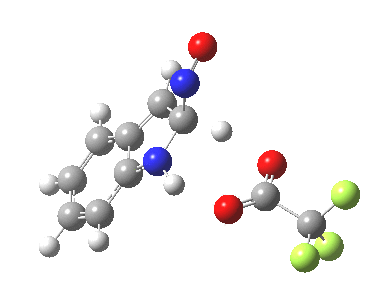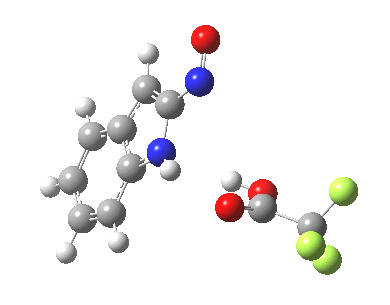
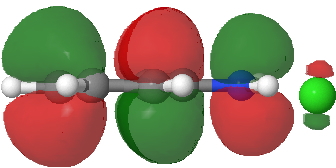
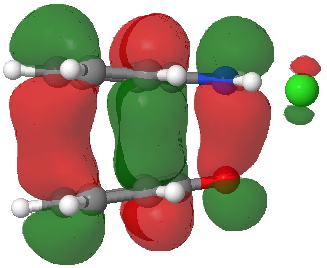
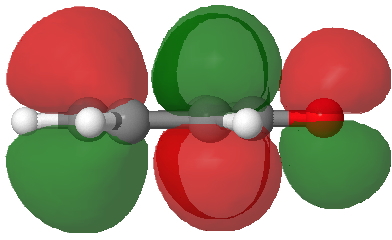
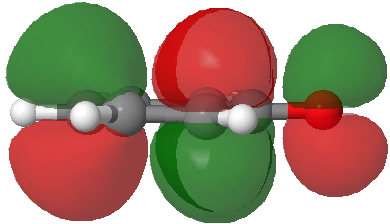
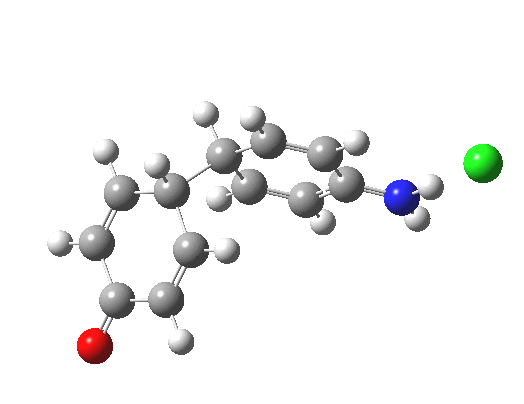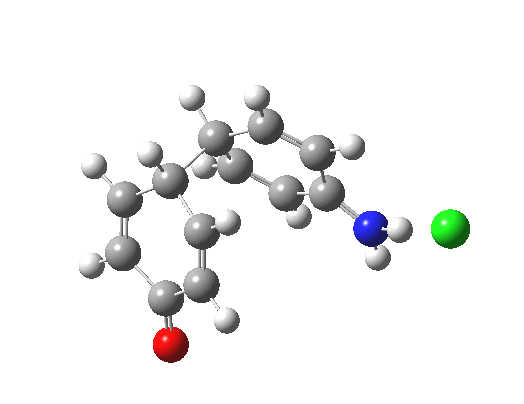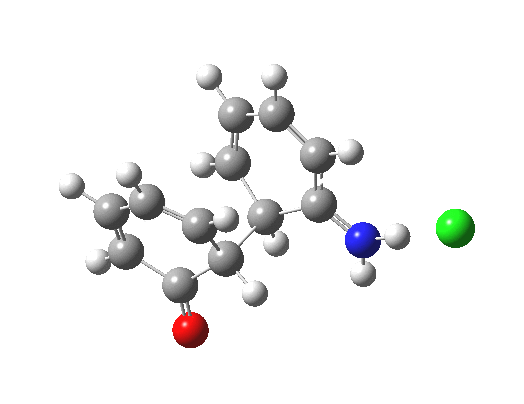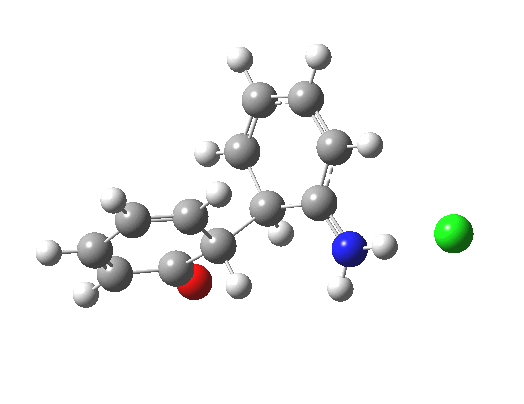
Back in the days (1893) when few compounds were known, new ones could end up being named after the discoverer. Thus Feist is known for the compound bearing his name; the 2,3 carboxylic acid of methylenecyclopropane (1, with Me replaced by CO2H). Compound 1 itself nowadays is used to calibrate chiroptical calculations[cite]10.1021/ct300359s[/cite], which is what brought it to my attention. But about four decades ago, and now largely forgotten, both 1 and the dicarboxylic acid were famous for the following rearrangement that gives a mixture of 2 and 3[cite]10.1021/ja00747a019[/cite]. I thought I might here unpick some of the wonderfully subtle stereochemical analysis that this little molecule became subjected to.
Feist’s acid and its derivatives have attracted constant attention a long while. The rearrangement shown above was identified in 1932, and by 1960 it was shown that 1 as a pure enantiomer gave products 2 and 3 that retained optical activity (read about all of this here[cite]10.1016/S0040-4020(01)92859-5[/cite]). By 1970 attention had shifted to the absolute configurations of the molecules involved and the mechanism of the reaction. Why? Woodward and Hoffmann had just put pericyclic reactions on the map[cite]10.1002/anie.196907811[/cite], and one of the examples they cited was this one. They identified the reaction as a [1,3]sigmatropic rearrangement (the red bond breaks and the blue bond forms) and their new theory required the configuration at carbon 1 to be inverted by the reaction, from (R) to (S) as shown above. In order to verify this, von Doering (who had been a student of Woodward’s) subjected Feist’s ester and its rearrangement products to a series of chemical transformations[cite]10.1002/anie.196907811[/cite] in order to relate its absolute stereochemistry to that of known compounds. Gajewski[cite]10.2012/ja00747a019[/cite] took over and with four further chemical transformations, was able to assert that the (S,S)-dimethyl enantiomer of 1 has an optical rotation of -59.4°.‡ The molecules 2 and 3 were subjected to a similar stereochemical analysis, which finally revealed them to have (S) configuration at the carbon labelled 1, thus confirming the inversion of configuration so confidently predicted by Woodward and Hoffmann. I imagine Feist never imagined the molecule which came to bear his name would be used as a confirmation of one of the pivotal 20th century stereochemical theories of organic chemistry.
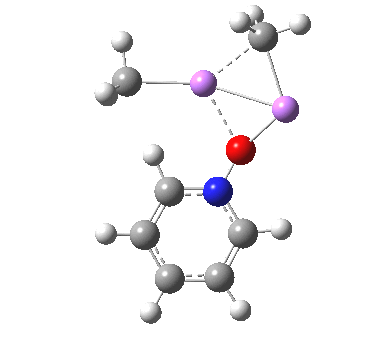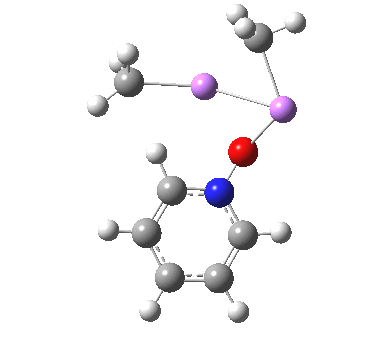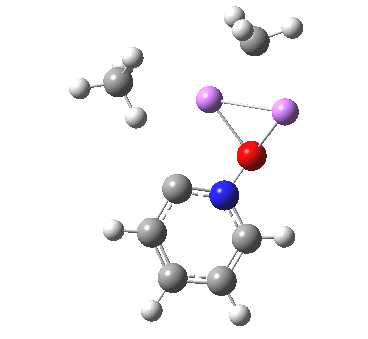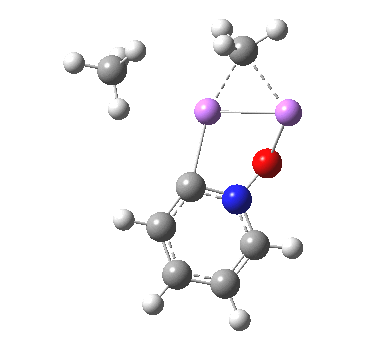
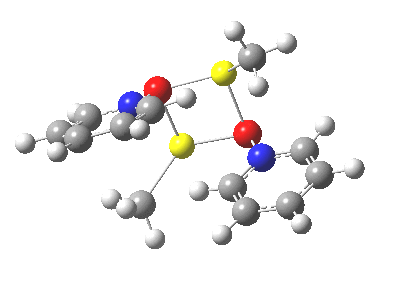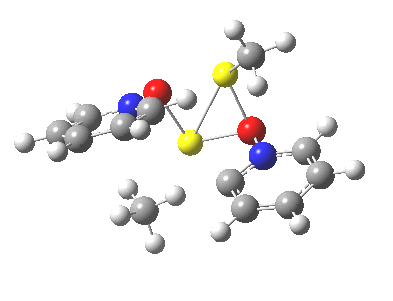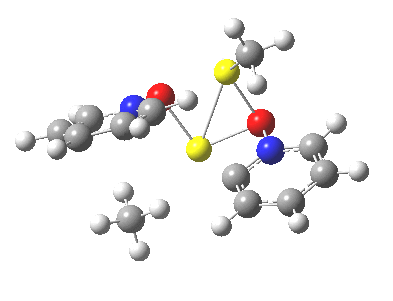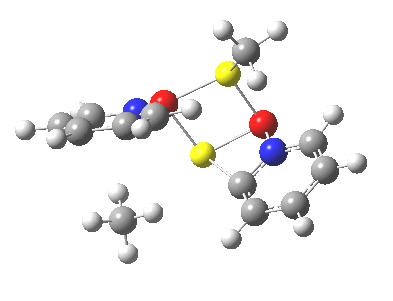
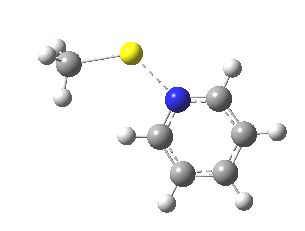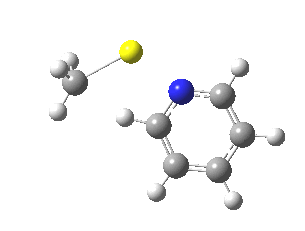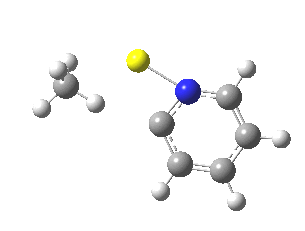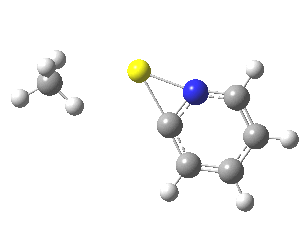
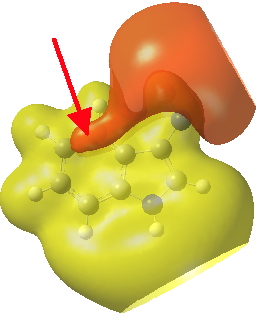
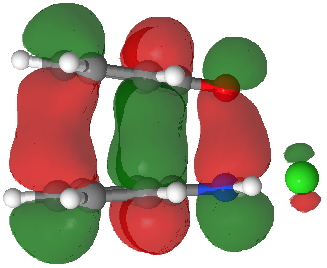
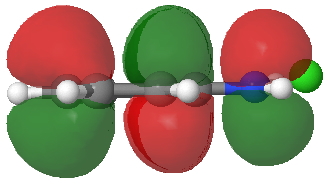
So what of the mechanism for this rearrangement? Well, a ωB97XD/6-311G(d,p) calculation reveals the transition state as shown below. The two dashed lines represent the red and blue bonds shown schematically above, and these bond either break or form to the same face of the three-carbon allyl fragment (suprafacially), but that carbon 1 (pointed to by the blue arrow below) suffers an Sn2-like inversion of configuration (= antarafacial) as proven by all that hard chemical synthesis noted above. 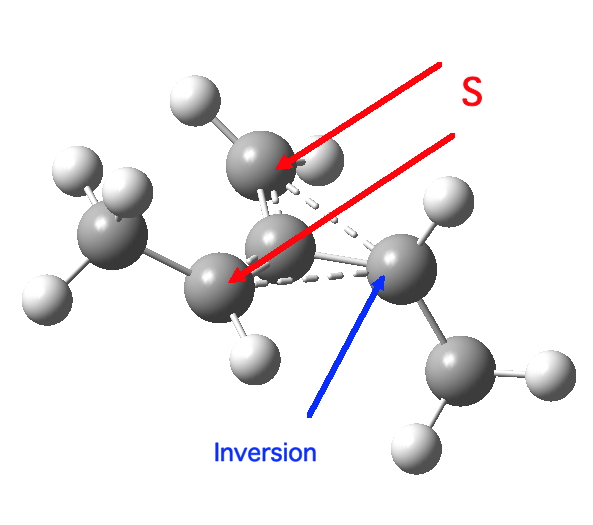
The reaction is concerted, with a predicted barrier of around 50 kcal/mol. This is a little higher than the measured value of ~41 kcal/mol[cite]10.1021/ja00901a009[/cite]. This is taken to indicate that the wavefunction has a contribution from an open-shell biradical configuration (indeed it is unstable at the transition state, having a lower energy triplet state) which would lower the barrier by 10-15 kcal/mol. The observation that the product has NOT lost optical activity suggests that the mechanism cannot simply be that of an achiral biradical, and that a “memory” of the starting stereochemical configuration must be retained throughout the dynamic reaction trajectory. Modelling such a process requires more sophisticated (multi-configuration) techniques than the one I have illustrated here, and quite probably a smattering of reaction dynamics thrown in. It goes to show that quite innocent looking molecules can be devils to model (both for their reaction dynamics and their optical activity!).
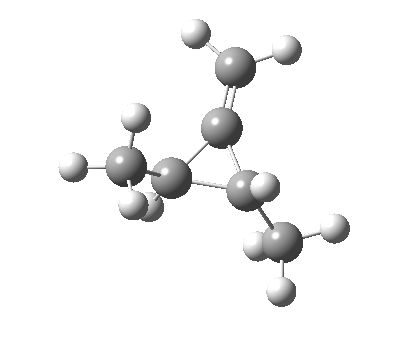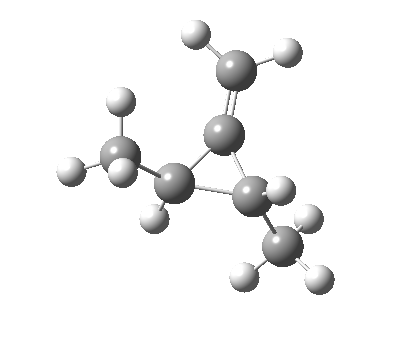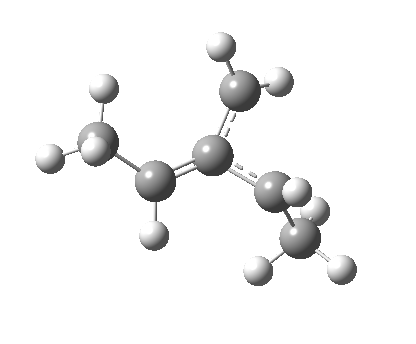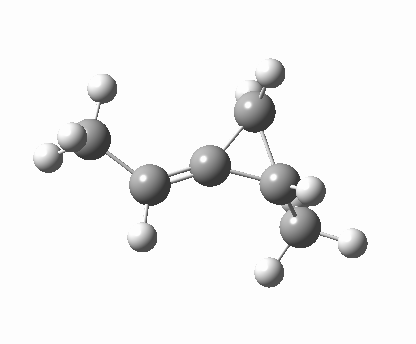 -[cite]10.6084/m9.figshare.670632[/cite] -[cite]10.6084/m9.figshare.670632[/cite] |
 |
Feist’s acid itself reveals a profile for the computed rearrangement IRC (ωB97XD/6-311G(d,p)/SCRF=water) that I have never seen as prominently before, a veritable table top of a mountain! This feature (and its reflection in the gradient norm) is a nice example of a “hidden intermediate”. In this case, it is a species which may be either biradical or zwitterionic, and which sits atop the mountain plateau. It can drop (bifurcate) off the mountain to form either compound 2 or 3, a process which must likely be best studied by dynamics rather than purely as an intrinsic reaction coordinates.
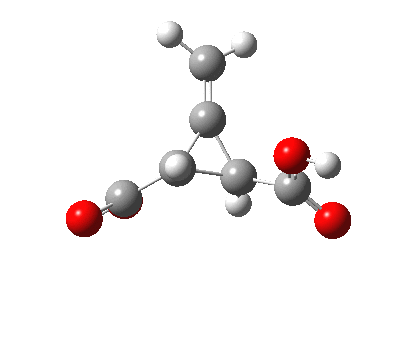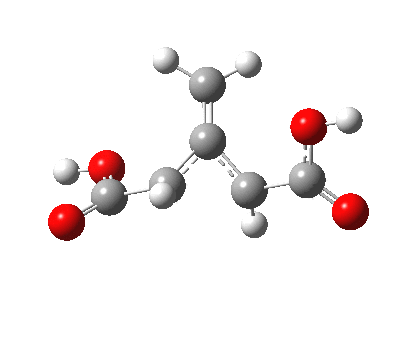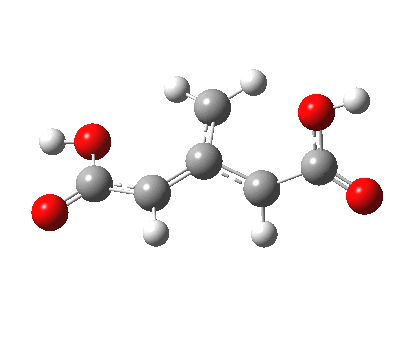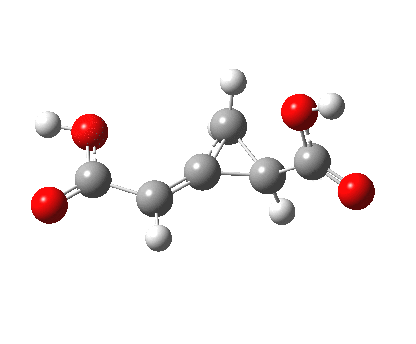 [cite]10.6084/m9.figshare.674600[/cite] |
 |
 |
|
‡See comment here.

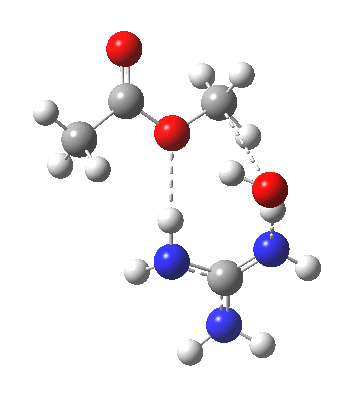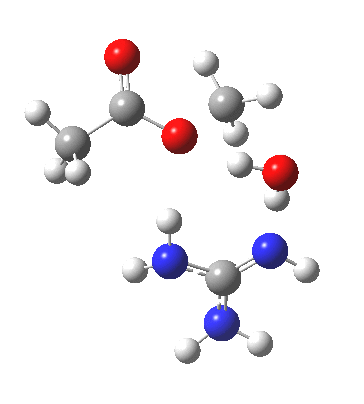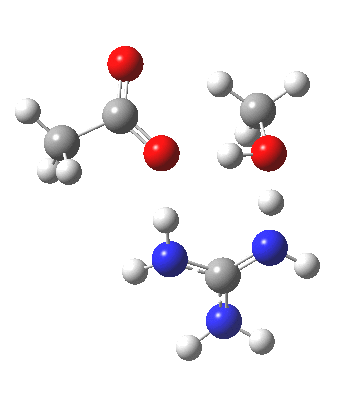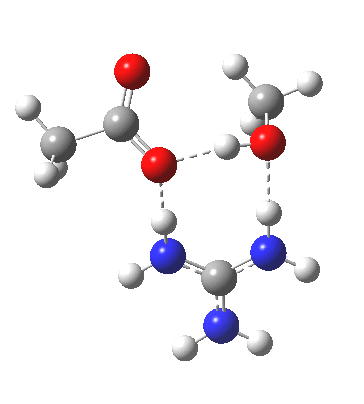 [cite]10.6084/m9.figshare.663603[/cite]
[cite]10.6084/m9.figshare.663603[/cite]
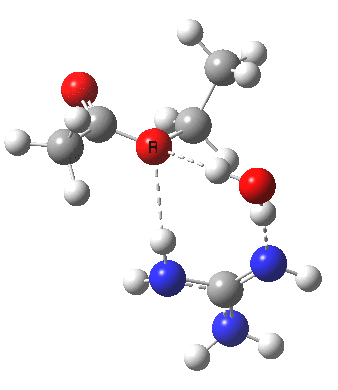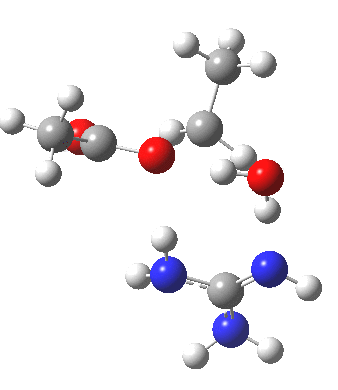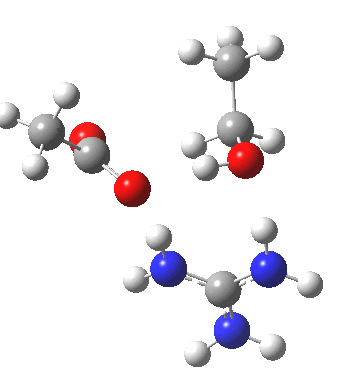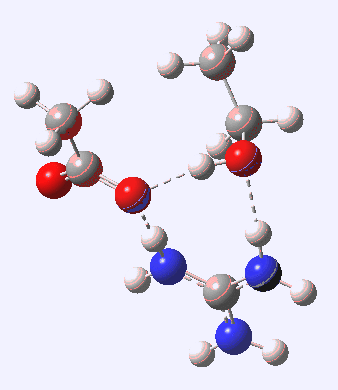 [cite]10.6084/m9.figshare.663619[/cite]
[cite]10.6084/m9.figshare.663619[/cite]

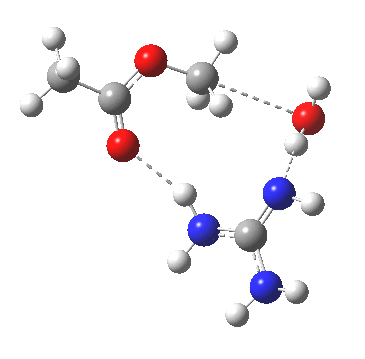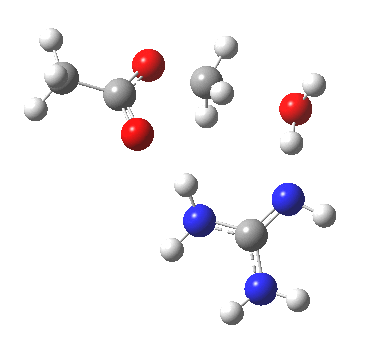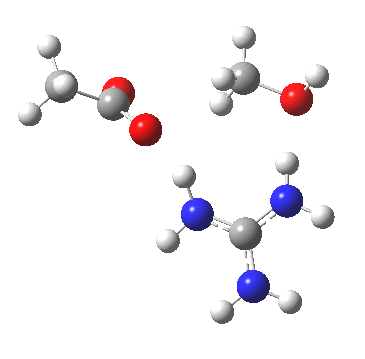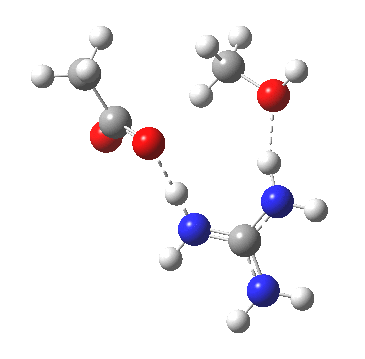


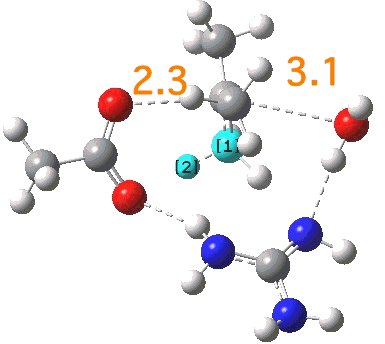


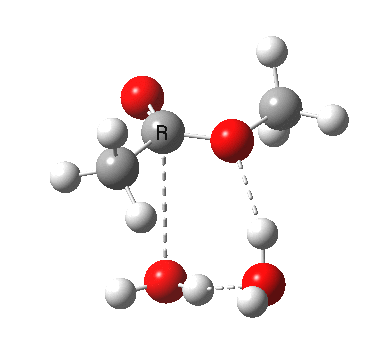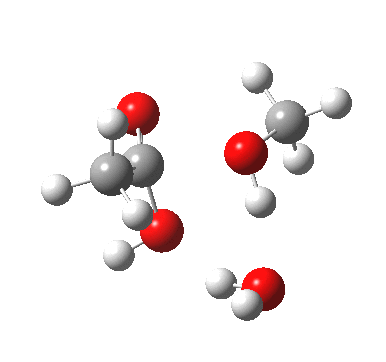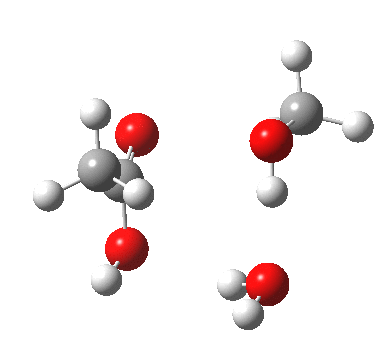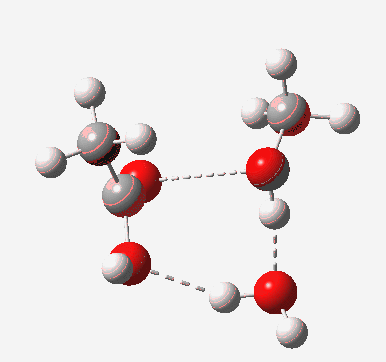 [cite]10.6084/m9.figshare.661351[/cite]
[cite]10.6084/m9.figshare.661351[/cite]
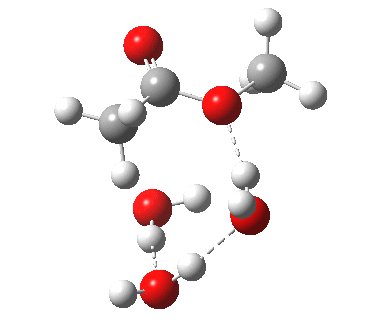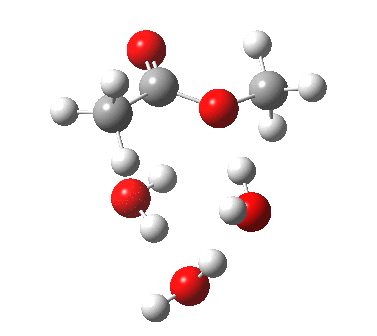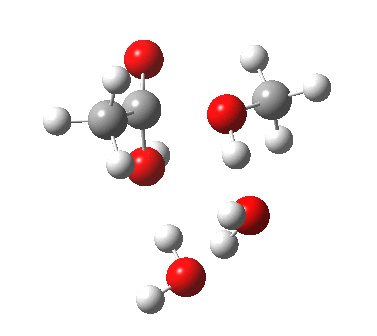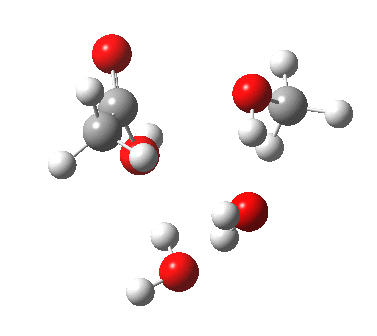 [cite]10.6084/m9.figshare.661789[/cite]
[cite]10.6084/m9.figshare.661789[/cite] 

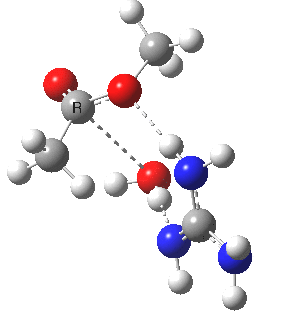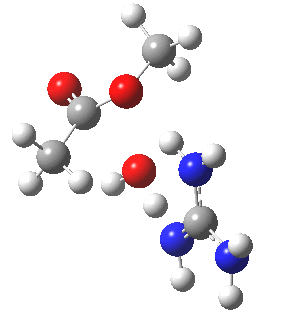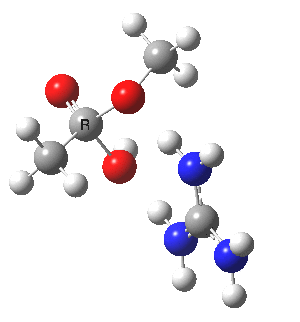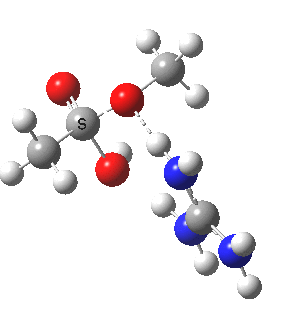 [cite]10.6084/m9.figshare.661791[/cite]
[cite]10.6084/m9.figshare.661791[/cite]
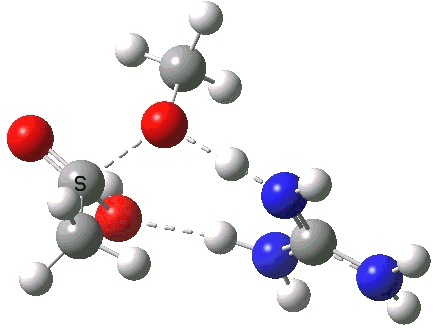

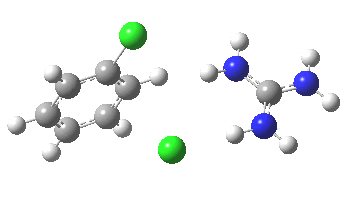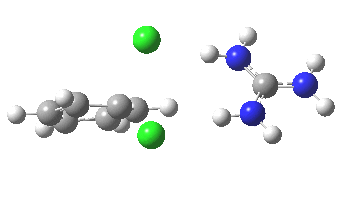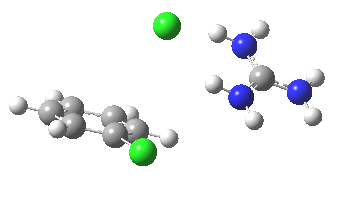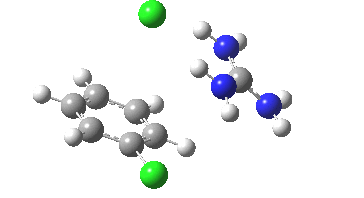 [cite]10.6084/m9.figshare.658893[/cite]
[cite]10.6084/m9.figshare.658893[/cite]
 [cite]10.6084/m9.figshare.658890[/cite]
[cite]10.6084/m9.figshare.658890[/cite]
 [cite]10.6084/m9.figshare.658910[/cite]
[cite]10.6084/m9.figshare.658910[/cite]
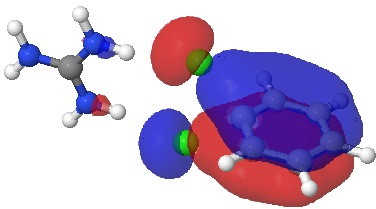
 [cite]10.6084/m9.figshare.659274[/cite]
[cite]10.6084/m9.figshare.659274[/cite]