Ever since the concept of a shared two-electron bond was conjured by Gilbert N. Lewis in 1916,[cite]10.1021/ja02261a002[/cite] chemists have been fascinated by the related concept of a bond order (the number of such bonds that two atoms can participate in, however a bond is defined) and pushing it ever higher for pairs of like-atoms. Lewis first showed in 1916[cite]10.1021/ja02261a002[/cite] how two carbon atoms could share two, four or six electrons to achieve a bond order of up to three. It took quite a few decades for this to be extended to four for carbon (and nitrogen) and that only with some measure of controversy and dispute (for one recent brief summary, see[cite]10.1039/D1CP02056K[/cite]).
For the transition elements over the last forty years or so, bond orders of four, five and even six between like atom pairs have been mooted and many characterised.[cite]10.1002/anie.201504414[/cite] Moving to the left of the transition elements in the periodic table, this hunt has looked at elements such as beryllium.‡ Eleven years back, I explored here how a Be=Be double bond could be formed, strangely enough as an electronically excited state of the dispersion-bound weak Be2 dimer.[cite]10.1139/v96-111[/cite] This species had a calculated Be-Be distance of 1.78Å, resulting from double excitation from the 2s σ*-antibonding orbital into the degenerate π-bonding orbital above it, giving four electrons in bonding valence orbitals. In 2019, three articles appeared which showed how this bond order might be extended to the lofty heights of three as in Be≡Be[cite]10.1002/cphc.201900051[/cite],[cite]10.1039/c9dt03321a[/cite],[cite]10.1016/j.cplett.2019.06.023[/cite] for (hypothetical) molecules in their ground electronic state. Here I discuss one example from these articles and compare it to the excited state observations made previously.
A useful starting point is the standard molecular orbital diagram for Be2, illustrating why the ground state singlet actually has a bond order of zero.

The three 2019 suggestions[cite]10.1002/cphc.201900051[/cite],[cite]10.1039/c9dt03321a[/cite],[cite]10.1016/j.cplett.2019.06.023[/cite] modified this to surround the Be2 core with e.g. six Li atoms, resulting in a stable singlet species with a Be-Be distance (calculated at e.g. the CCSD/Def2-TZVP level) of 1.99Å and exhibiting C2h symmetry.♥ The role of the Li is to polarise and repopulate Be orbitals by delocalization of e.g. a 2c-2e bond in Be2 dimer into a 6c-2e bond in Be2Li6. The reported calculations (as successfully replicated here†, FAIR DOI: 10.14469/hpc/10106) show the resulting molecular orbitals for Be2Li6 comprise an (accidentally) degenerate π-pair and a higher energy weak σ-orbital, together forming the proposed triple bond. This of course inverts the normal ordering of such bonds, for which the σ-orbital is lower in energy (more stable) than π-bonds. The form of the σ-orbital also reminds to some extent of the fourth σ-bond in C⩸C.
| MOs for Be2Li6 |
|
HOMO, σ orbital
-0.158au
|
HOMO-1, π-pair,
-0.175au
|
HOMO-2, π-pair
-0.176au
|
 |
 |
 |
| Because the static 2D projections shown in the articles cited above do not always make for easy interpretation, if you click on the orbital thumbnails, you will get dynamic 3D isosurfaces to rotate and inspect. These were generated using the tool at https://www.ch.ic.ac.uk/rzepa/cub2jvxl/ |
The two lower energy 2s σ-orbitals, which taken together do not apparently contribute to the overall bond order in Be2Li6, are shown below.
| Lower energy MOs for Be2Li6 |
| σ -0.235au |
σ-0.496au |
 |
 |
ELF (electron localisation function) integrations for Be2Li6 show each beryllium has two basins in the Be-Be region of about 2.5e each (red arrows) typical of triple bonds and two terminal Li-Be basins of 2.3e.

One aspect arising from my earlier post on the excited state Be=Be double bond relates to the reported calculated Be-Be bond length of 1.99Å and ν 718 cm-1 for ground state Be2Li6. To quote one article[cite]10.1002/cphc.201900051[/cite], “the Be≡Be triple bond in Li6Be2 may also be considered as another example of an ultraweak but ultrashort triple bond.” I had noted earlier that the electronically excited state of the Be2 dimer has a computed bond length of 1.78Å and ν 917 cm-1 for a double bond order, this being significantly shorter than the suggested ultrashort triple bond. We learn from this that the relationship between a bond order and a bond length may not always be linear. In other words, a longer bond may in fact have a higher bond order than a shorter bond between the same two atoms. The same was true as it happens with C⩸C; the mooted quadruple bond had a longer bond length than the triple bond in HC≡CH. That observation was controversial at the time; I suspect a similar phenomenon for Be has become less controversial.
To go back to the Be=Be dimer which started things off and that excited state with one electron in each of the degenerate π-orbitals (actually a triplet state). What would happen if two electrons were to be added, making an excited state of Be22-? Yes indeed, this species (CCSD/Def2-TZVPPD) has a calculated bond length of 1.885Å and ν 766 cm-1. If this di-anion is stabilised with a continuum water field (a milder version of surrounding the dimer with Li atoms), the Be-Be length contracts to 1.74Å, the Be-Be stretch increases to 949 cm-1 and the σ-orbital becomes more stable than the π-orbitals. At the higher CCSD(T)/Def2-TZVPPD/SCRF=water level, the bond length still has the ultrashort value of 1.761Å, which might be assumed as the natural value for Be≡Be, a classical triple bond. From that perspective, the “ultraweak but ultrashort triple bond” predicted for Be2Li6 actually emerges as a relatively long triple bond!
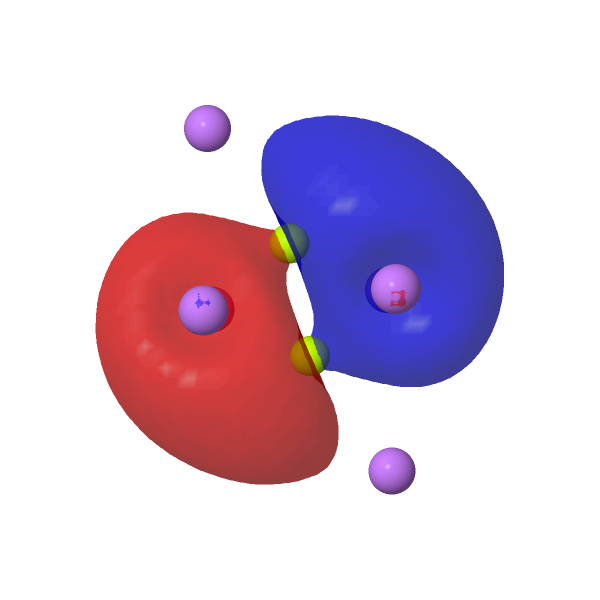
Our final exploration is to add two lithium atoms to Be2 to form the neutral LiBe≡BeLi. This was done in stages (see FAIR DOI 10.14469/hpc/10106), starting with a linear arrangement of atoms which revealed two negative force constants, a C2h shape with one negative force constant and ending with a C2 (chiral!) geometry with no negative force constants. This has a Be≡Be length of 1.705Å (ωB97XD/Def2-TZVPPD/SCRF=water), ν 1129 cm-1, a Wiberg bond index of 2.98 and a Li-Be bond index of 0.0065, indicating an entirely ionic lithium and again a central Be22- unit. As an excited state, it is 49.8 kcal/mol higher than the ground state of Be2Li2.
| NBOs for LiBe≡BeLi |
|
HOMO, π-pair,
-0.175au
|
HOMO, π-pair
-0.176au
|
HOMO-2, σ orbital
-0.158au
|
 |
 |
 |
So to conclude, we have seen two different motifs for constructing a model of a Be≡Be triple bond, one recently reported in the literature for a ground state species with six lithium atoms surrounding the Be2 dimer and a simpler one with just two lithiums exhibiting a much shorter Be≡Be bond but which requires electronic excitation to achieve. So these two motifs are not equivalent. But hopefully this exercise shows how playing around with atoms and electrons can achieve very unusual bonding states and elevated bond orders from which one can learn a lot, although with the caveat that one does not always produce molecules capable of facile synthesis!
‡On a slightly different theme, Cs can be shown to sustain three bonds, albeit all to different atoms. See DOI: 10.6084/m9.figshare.861030 Li≡Li4- can also be calculated as the tetra-anion showing almost identical properties to Be≡Be2- with a Li≡Li triple bond distance of 2.11Å. See DOI: 10.14469/hpc/10122. †Replication was necessary because the appropriate wavefunction files for analysis were not included in the supporting information. Only the coordinates were available for interoperation, and due to a quirk in the way Adobe Acrobat works, even those could not be easily transferred by a simple copy/paste operation to create a job input file. See e.g. here or DOI: 10.14469/hpc/10043 for more discussion. All the wavefunction files for this replication are available at the FAIR DOI noted above. ♥The Be-Be distance in catena(dimethylberyllium), a polymer comprising two bridging Me units connecting Be atoms, is only slightly longer at 2.09Å[cite]10.1107/S0365110X51001100[/cite] This fascinating transannular Be-Be interaction is one to be explored elsewhere.
The post has DOI: 10.14469/hpc/10125




























{kind=link}
{kind=link}