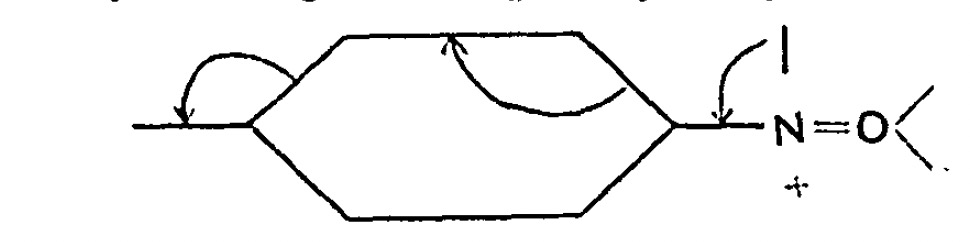
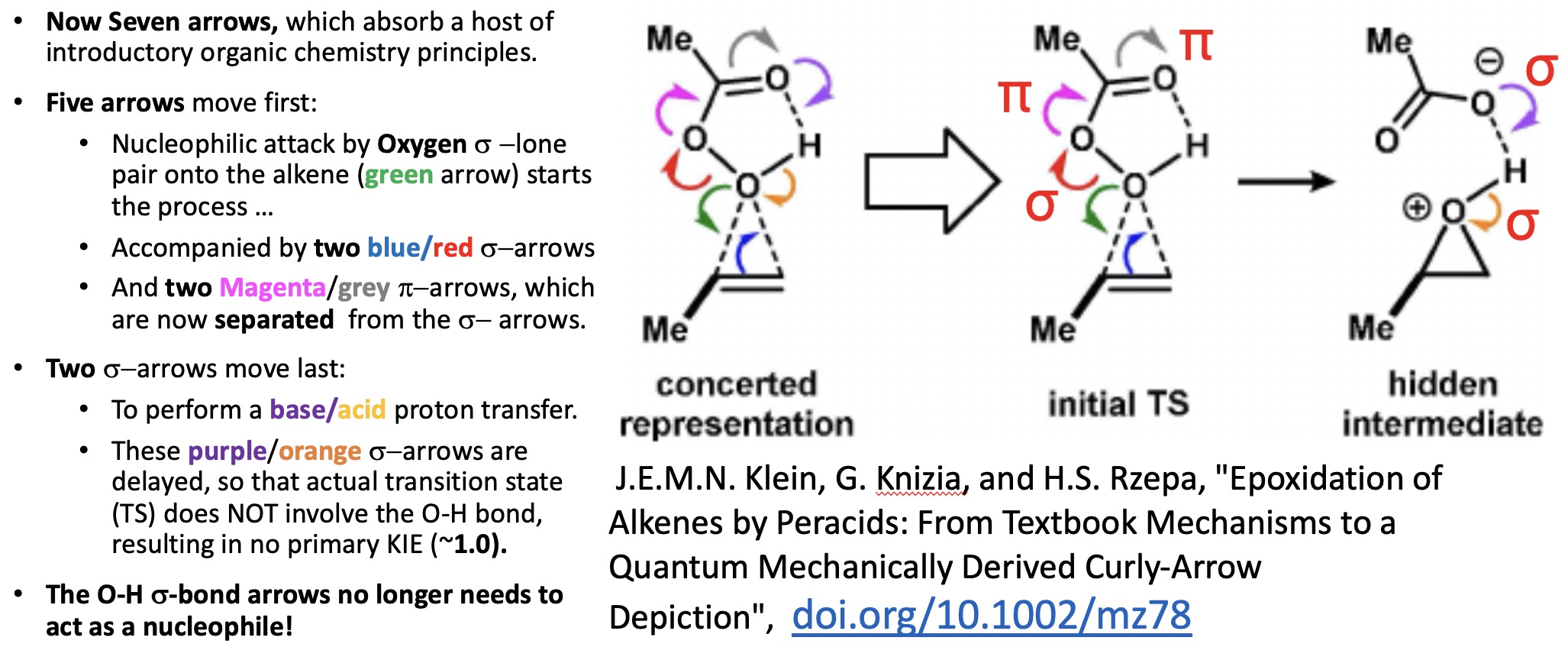
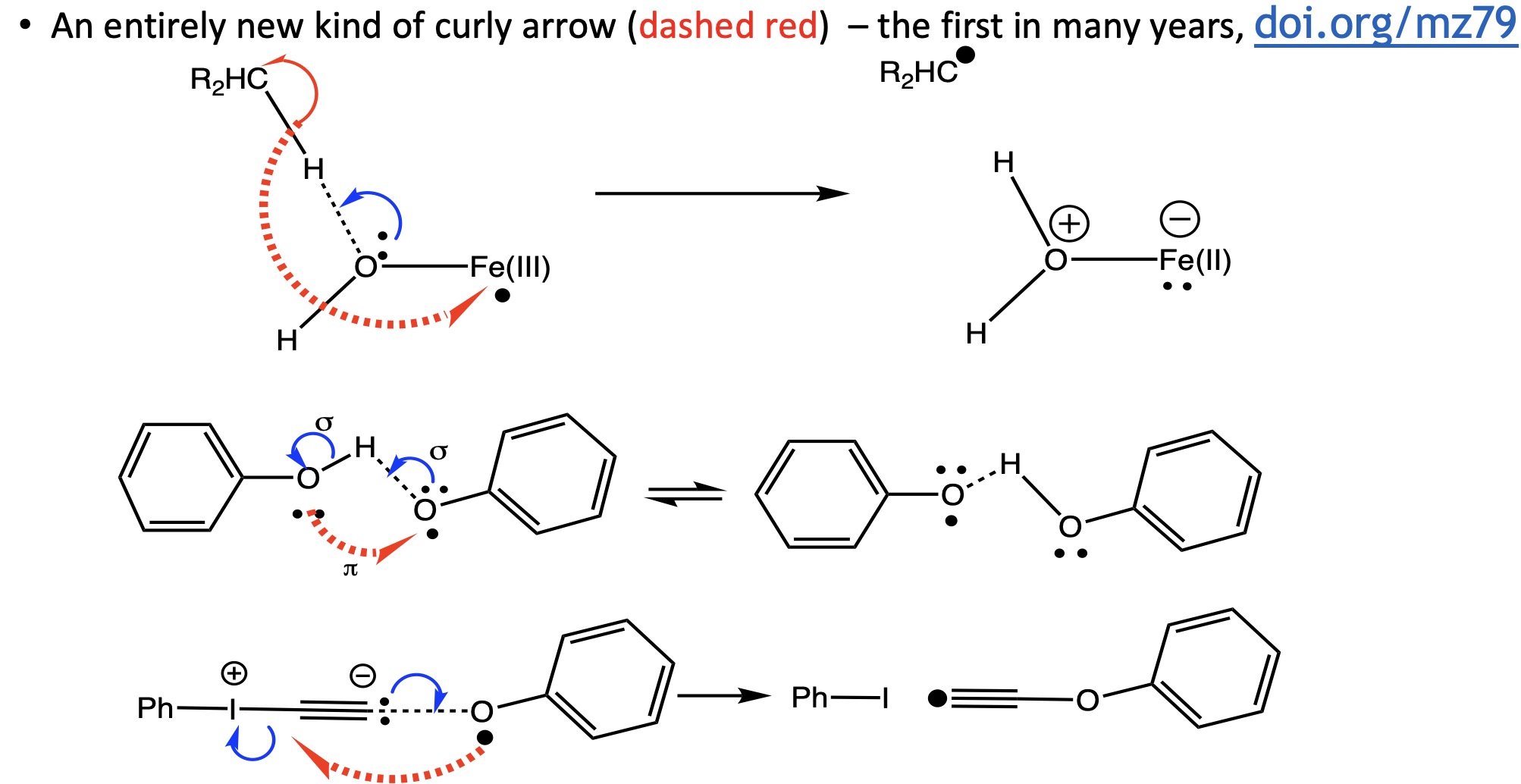
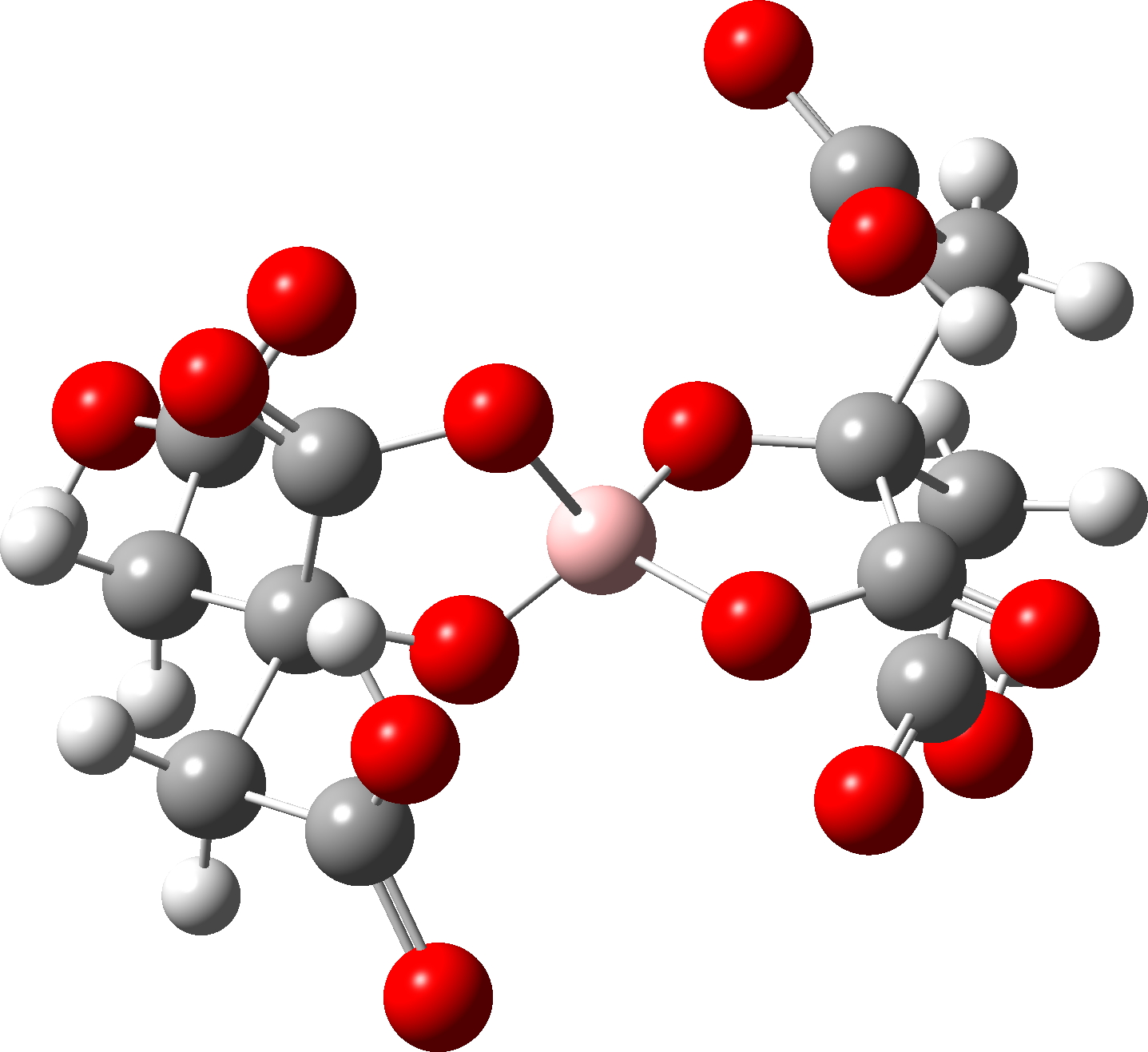
I have written a few times about the so-called “anomeric effect“, which relates to stereoelectronic interactions in molecules such as sugars bearing a tetrahedral carbon atom with at least two oxygen substituents. The effect can be detected when the two C-O bond lengths in such molecules are inspected, most obviously when one of these bonds has a very different length from the other. The effect originates when one of the lone pair of electrons on one oxygen atom uniquely overlaps with the C-O antibonding σ* on another oxygen, thus shortening the length of the donating oxygen-carbon length and lengthening the length of accepting C-O bond. Here I take a look at tetra-substituted versions of this (C(OR)4), where in theory there are up to eight lone pairs, interacting with any of three C-O bonds, giving a total of 24 possible anomeric effects in one molecule.

We start the process with a search of the Cambridge crystal structure database, using the following search query:

This yields 25 hits. We now want to find out what the longest and shortest C-O bonds are, and how large the difference between them is. To do this, we have to resort to applying some functions, using the calculator tool built into the Mercury analysis software. The following functions were used:
- Greatest('search3'.'DIST1','search3'.'DIST2','search3'.'DIST3','search3'.'DIST4')
- Least('search3'.'DIST1','search3'.'DIST2','search3'.'DIST3','search3'.'DIST4')
- Greatest('search3'.'DIST1', 'search3'.'DIST2', 'search3'.'DIST3', 'search3'.'DIST4')-Least('search3'.'DIST1', 'search3'.'DIST2', 'search3'.'DIST3', 'search3'.'DIST4')


The results can be displayed as below, in which the difference between the two bond lengths is colour coded (red = greatest, blue = least).

- Here you can see that when the difference between the longest and short C-O bond lengths is small, the colour is blue.
- Green dots show a difference of about 0.04-0.05Å
- The red dot has the greatest difference of 0.087Å and corresponds to the entry SILDOH ([cite]10.1107/S1600536807042298[/cite], DataDOI: [cite]10.5517/ccq8lq8[/cite], 10.5517/ccq8lq8.
The next step is to apply a “reality check” using computation, here a MN15L/Def2-TZVPP calculation on the top eight entries as sorted by the largest C-O bond length differences (ΔrC-O > 0.05Å.[cite]10.14469/hpc/13925[/cite], data DOI: 10.14469/hpc/13925
| CCDC Ref code |
Crystal structure |
Computational structure |
| Longest |
Shortest |
Δ |
Longest |
shortest |
Δ |
| SILDOH |
1.451 |
1.364 |
0.087 |
1.441 |
1.367 |
0.074 |
| PILTOU |
1.432 |
1.361 |
0.071 |
1.418 |
1.378 |
0.040 |
| GISSAD |
1.435 |
1.367 |
0.068 |
1.422 |
1.375 |
0.047 |
| BODGEG |
1.507 |
1.442 |
0.065 |
1.424 |
1.370 |
0.054 |
| GINLOF |
1.425 |
1.364 |
0.061 |
1.418 |
1.377 |
0.041 |
| POCPOO |
1.419 |
1.361 |
0.058 |
1.421 |
1.371 |
0.050 |
| KEVFUM |
1.417 |
1.361 |
0.056 |
1.395 |
1.391 |
0.004 |
| AHEYAO |
1.423 |
1.370 |
0.053 |
1.422 |
1.372 |
0.050 |
- The largest effect occurs for SILDOH, and this is replicated by calculation.
- The largest discrepancy between measurement and calculation is for KEVFUM, where calculation predicts almost no C-O bond differences. This will be discussed elsewhere.
Focusing on SILDOH, we look at the NBO E(2) energies for the donor-acceptor interactions of an oxygen lone pair donating into a C-O antibonding σ* orbital.

Click on the image below for a 3D model of the two interacting orbitals (positive overlap = blue + purple, red + orange)

The interaction of LpO1 to the long bond C5-O4 = 18.0 and LpO2 to C5-O4 = 16.3 kcal/mol, whereas in the reverse directions, LpO4 to C5-O1 is only 6.0 kcal/mol and LpO4 to C5-O2 is 10.7 kcal/mol. For a “normal” C-O bond however such as C5-O3, LpO2 to C5-O3 = 3.1 and LPO1 to C5-O3 = 5.3 kcal/mol. In effect, two oxygens “gang up” on weakening the long C5-O4 bond, but leave the shorter C5-O3 bond alone. So the individual anomeric effects are no larger than normal, but the cooperative effect of two acting together is what produces the final geometric asymmetry.
The Wiberg bond index mirrors this effect. The bond indices are 0.9882 for O1-C5 and C5-O4 0.8512 (Δ =-0.137) which is a big difference in bond order and accounting for the large (record?) difference in bond length.
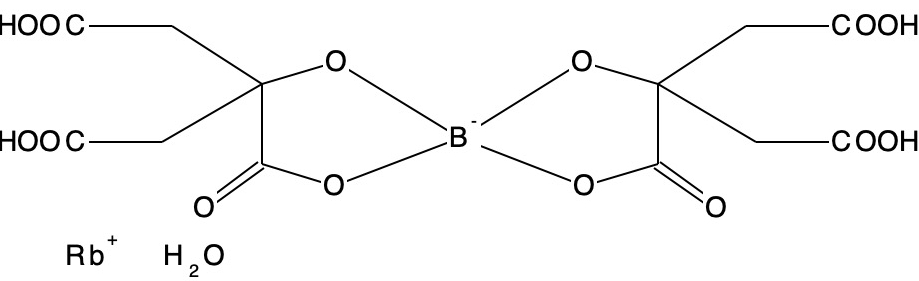
In the next post, I will analyse the equivalent molecules B(OR)4–.