Some 13 years ago, I speculated about the longevity of the type of science communication then (and still now) represented by Blogs. I noted one new project called ArchivePress that was looking into providing solutions equivalent to what scientific journals have done for some 350 years of science communication. The link to ArchivePress no longer works, but details of the project can still be found here. Since then the technology and infrastructure has moved on, with a new backbone provided by the use of persistent identifiers (PIDs) in the form of DOIs. The PID ecosystem is now extensive and so a revival of the concept has recently been launched called The Rogue Scholar. Here I take a look at some of is features and illustrate these with application to this blog.

To quote its aims “The Rogue Scholar improves your science blog in important ways, including full-text search, long-term archiving, DOIs and metadata”. Lets take these four ways and compare them with how scientific journals function.
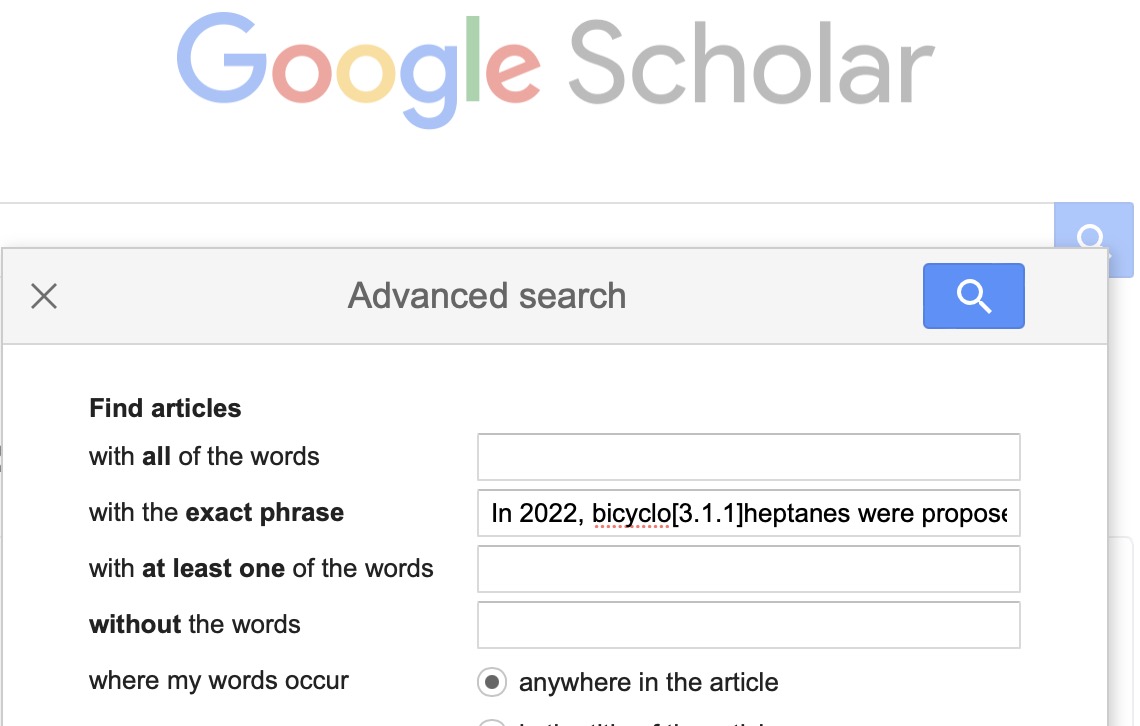
- Full text search. The traditional journal is full-text indexed by its publisher, but of course there are many science publishers out there and they focus on indexing only their own journals. There are an estimated 30,000+ science journals, covered by commercial abstracting agencies such as SciFinder. A search engine that aggregates full text searches across (most?) journals is Google of course. Type a full text search string, enclosing it in quotes to get a literal search of the entire string,‡ and you are quite likely to find the journal article. Try this for yourself and report back if it does not work. If you try scholar.google.com instead, it will not work (even using the Advanced search/Exact phrase constraint). Try then some text from a blog† and again with scholar.google.com it will not work but with Google it does (I have not tried other search engines).
-
- Now try eg the blog search using RogueScholar and you get the successful result shown below.

- Full “advanced mode” searching is of course second nature to chemists, who apply a variety of fielded or constrained searches are part of their regular routine (including eg chemical substructure searching), but you often need specialist abstracting and indexing agencies such as Scifinder for this. But RogueScholar will offer constrained searches I understand at some stage, based on “metadata” and so I will take this topic next.
- Now try eg the blog search using RogueScholar and you get the successful result shown below.
-
- MetaData and DOIs. When a journal article is published, part of the process is to gather metadata about the article and submit it to a metadata aggregating agency such as CrossRef. In exchange, the latter offer a DOI which functions as a unique and persistent identifier for that article. The metadata terms can be used in a constrained search with CrossRef, and with DataCite for FAIR data. RogueScholar does exactly the same, it gathers metadata from a registered blog post, including ORCID identifiers associated with the post, registers it and enables searching. Try e.g. 0000-0003-3315-3524 as a search term and see what you get. This also works for DOIs (try 10.59350/65c45-3ew97 as a search term). So here too, we can see parity emerging with conventional journal publishing.
- The final component is long-term archiving. Again journals have been doing this for a long time (on paper) and in the last 30 years or so in digital form. Blogs have until now lacked this and here again RogueScholar is promising such long term archiving in its next iteration.
- This actually raises one interesting, albeit difficult, aspect of blogs – one however that may in fact be somewhat unique to this particular blog. The topic of my very first post here back in 2008 was how interactive 3D molecular models could be included in the post itself to help the reader explore the chemical points I was trying to make. Back then I regarded it as something that could not be so easily done as part of journal articles[cite]10.1080/13614579509516846[/cite] (although you do see it nowadays), but that feature certainly presents a challenge to long term archival! I do not see a solution for this one on the horizon.
Rogue Scholar is still a very young service, and no doubt will evolve rapidly from this point, so I may revisit in say six months time to see how it has come along. Meanwhile, try it out and see what you think. And because science blogs can now be assigned a DOI on the same level as journal articles, they too can join the so-called universe of “Knowledge or PID graphs“[cite]10.1016/j.patter.2020.100180[/cite].
‡ For example “In 2022, bicyclo[3.1.1]heptanes were proposed to mimic the fragment of meta-substituted benzenes in biologically active compounds”
† “The schematic representation of a chemical reaction mechanism is often drawn using a palette of arrows connecting or annotating the various molecular structures involved. These can be selected from a chemical arrows palette, taken for this purpose from the commonly used structure drawing program Chemdraw.”
This post has DOI: 10.59350/8m2d8-47b52













 But now observe the bond length response, which shows a distinct maximum around the first transition state (IRC ~7). This is the opposite behaviour to the previous two systems, and now indeed matches the original inferences one might make from the double headed arrow.
But now observe the bond length response, which shows a distinct maximum around the first transition state (IRC ~7). This is the opposite behaviour to the previous two systems, and now indeed matches the original inferences one might make from the double headed arrow.















 Here I take a look at this very unusual structure, the core of which is an imidazole ring coordinated via nitrogen to the metal Cu.
Here I take a look at this very unusual structure, the core of which is an imidazole ring coordinated via nitrogen to the metal Cu.
 reveals 12 hits, with a range of O-O distances ranging from 1.37 to 1.54Å. A histogram of the O-O lengths in the Cu(O-O)Cu sub structure shown below shows quite a distribution amongst the 12 known examples.
reveals 12 hits, with a range of O-O distances ranging from 1.37 to 1.54Å. A histogram of the O-O lengths in the Cu(O-O)Cu sub structure shown below shows quite a distribution amongst the 12 known examples.