The Royal Society of Chemistry historical group (of which I am a member) organises two or three one day meetings a year. Yesterday the October meeting covered (amongst other themes) the fascinating history of madder and its approximately synthetic equivalent alizarin. Here I add a little to the talk given by Alan Dronsfield on the synthesis of alizarin and the impact this had on the entire industry.
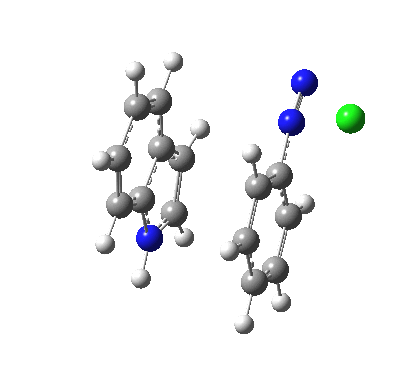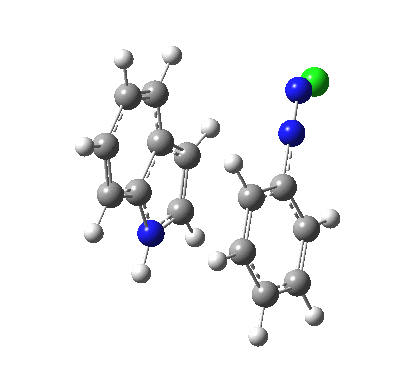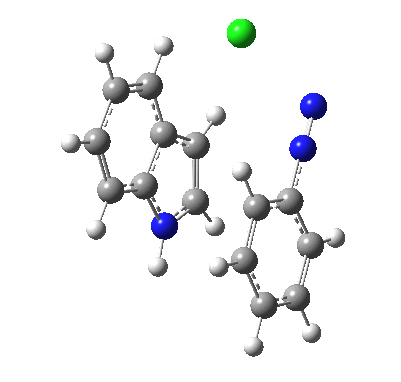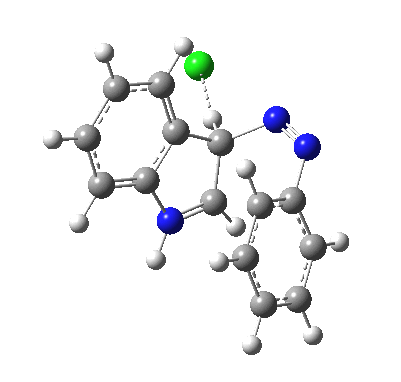
Although William Perkin famously (and accidentally) produced the first synthetic chemical dye in 1856 (Mauveine), the industry at that time was both large and dominated by dyes from natural products. Mauve was something of a niche colour; far more important was alizarin, both as a red dye (for cotton) and a red pigment (in painting) and up to 1869 it was sourced from the roots of the madder plant (which was difficult to farm) and from insects (which could be farmed). It was nonetheless expensive to produce it from either and so a race started to create it synthetically. Famously, two groups submitted patents for such a synthesis in 1869, William Perkin himself and two scientists working in BASF, Carl Graebe and Carl Liebermann.[cite]10.1002/cber.18690020106[/cite],[cite]10.1002/cber.186900201141[/cite] The latter were the winners (by one day) and they are now famed for their work‡ (what a difference one day can make; Perkin is known for his other work, but not as much for the synthesis of alizarin). As with mauveine, the structures of these dyes were not known with certainty (or for mauveine even approximately) at the time, but Graebe and Liebermann had managed to prove that alizarin was derived from anthracene by reducing the former to the latter using zinc dust. Trouble was, the structure of anthracene itself was not certain in 1869! There were two probable candidates, (a) and (b) below.

Alan told us how Graebe and Liebermann favoured structure (a), now known as phenanthrene, rather than (b), which we recognize as anthracene. A full story is told in this PhD thesis, written in 1919 and published in 1921[cite]10.1021/ja01437a023[/cite] and I can only tell a tiny bit of it here. Essentially (a) was preferred over (b) because the former could sustain three aromatic (benzene-like) rings, whereas the latter only two (p 3 of the thesis above). Years later in 1972, this concept emerged as the Clar π-sextet rule, but the idea was already more than 100 years old by then! And indeed thermodynamically, phenanthrene is more stable than anthracene. By 1872, circumstantial evidence was accumulating that in fact alizarin was derived from (b), largely via attempts to synthesize the molecule by various reactions. These often were performed at high temperatures (red-hot tubes), and we now know that many complex rearrangements can occur at such temperatures. In 1889[cite]10.1039/pl8900600095[/cite], Armstrong was quoting the structure of anthracene with no doubts about its structure. However, it took another 30 years or so for an entirely unambiguous total synthesis of anthracene to be devised.[cite]10.1021/ja01437a023[/cite] Also around that time the first structures based on crystallography were emerging (by William Bragg) that supported this hypothesis. Even so, the first modern crystal structure had to wait until 1950.[cite]10.1107/S0365110X50000641[/cite]
We learn from this story that many chemical structures established during the 19th century were largely based on (admittedly a large) body of circumstantial evidence. A wonderful example of how a systematic rather than a circumstantial proof of the structure of naphthalene was established using chemical synthesis and degradations alone can be found here in the work by Armstrong. Evidence obtained from instruments was largely restricted to techniques such as thermochemistry and polarimetry in the 19th century and for the first twenty years of the 20th to e.g. infra-red spectroscopy.[cite]10.1103/PhysRevSeriesI.20.273[/cite] It is remarkable then that actually, most 19th century structures have stood the test of time. Moreover, not knowing the precise structure did not prevent the processes for making them to be patented. Nowadays of course, a simple crystal structure can often be solved in a few minutes and NMR spectroscopy takes a similar amount of time. We are no longer used to waiting for years or indeed decades for structural proof!
‡This synthesis proved to be very expensive (requiring a step using bromine and then a second step to remove it). But shortly after, a much more efficient synthesis which dispensed with the bromine brought the cost of the dye down dramatically. The madder industry never really recovered from this blow.

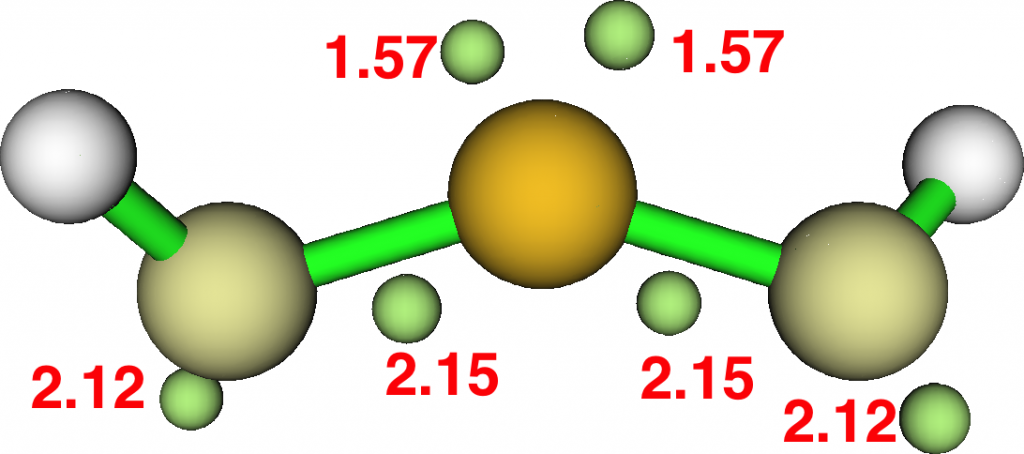
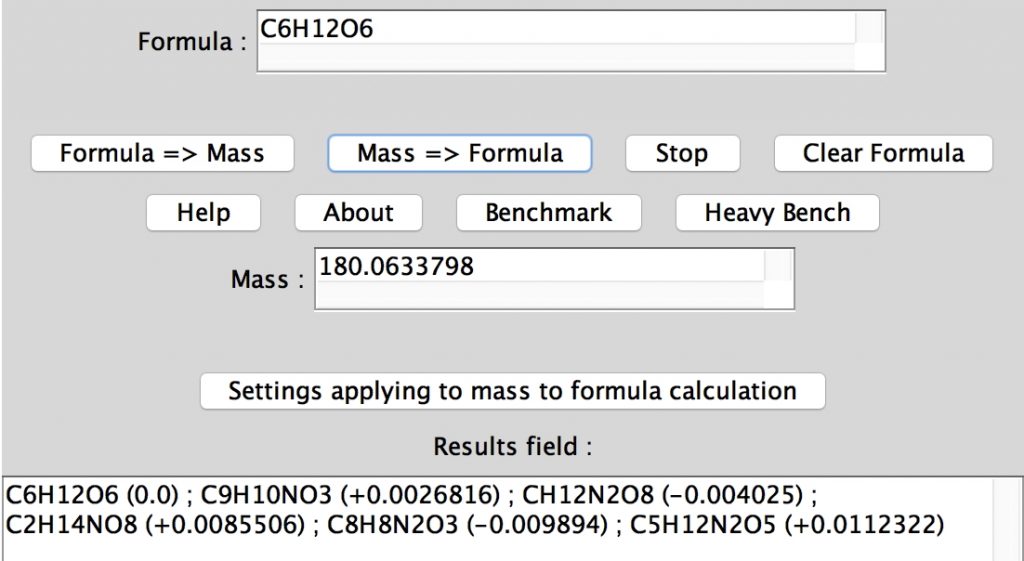 The applet also has a benchmark feature. Running the heavy bench now takes ~ 0.4s on a laptop. I cannot be sure, but I seem to remember that this one took ~20 seconds back in 1997.
The applet also has a benchmark feature. Running the heavy bench now takes ~ 0.4s on a laptop. I cannot be sure, but I seem to remember that this one took ~20 seconds back in 1997. 




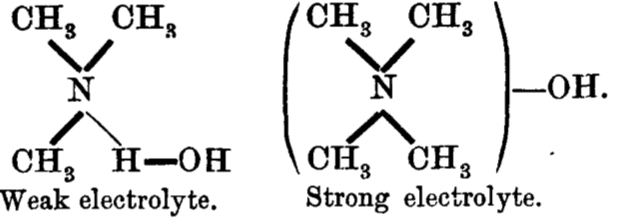

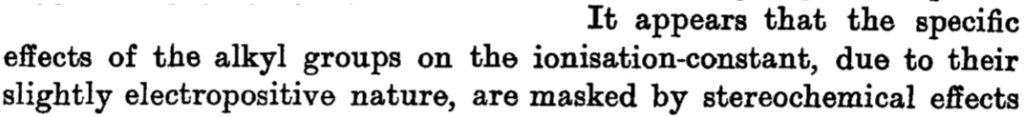
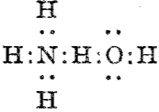
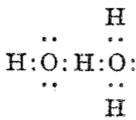








 The main points of this argument were;
The main points of this argument were;