Continuing my european visits, here are two photos from Bonn. First, a word about how the representation of benzene evolved, attributed to Kekulé.
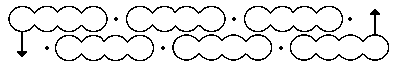
Above is his first effort, made in 1865.
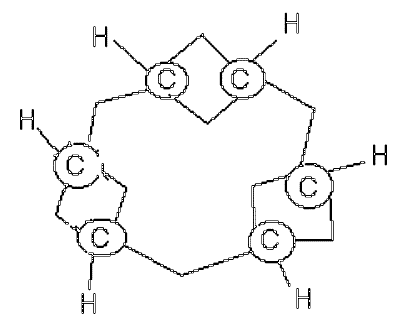
This one above is better, offered in 1866. But whilst what we now know as the double bond (C=C) is perhaps understandably kinked (and we now call these banana bonds, since nature tends to abhor kinks in electron density), so too are the single bonds!

So when it came to erecting a statue in his honour around 1890, the kinks were straightened out! The figure on the right (female) represents science (and purity). The two chaps on the left are workers representing industry. Note that they have still not quite gotten the lengths of the putative single and double bonds in proportion. By 1872 of course, Kekulé had proposed[cite]10.1002/jlac.18721620110[/cite],[cite]10.1002/jlac.18721620211[/cite] his oscillating model, the one that is taught to this day.
I feel I should add one modern interpretation to this concept. An oscillation implies a frequency. Kekulé could only know that this oscillation was fast on what might be called the “laboratory scale” (in other words, no-one had been able to isolate the individual isomers of substituted benzenes, which implies that the rate constant inter-converting them was probably faster than k = 10-3 s-1 or a few minutes half-life).‡ We now know that this oscillation is ~1014 s-1, the timescale of a molecular vibration! By the way, transition state theory tells us that Ln(k/T) = 23.76 – ΔG‡/RT where k is the unimolecular rate constant, ΔG‡ the free energy barrier, T the temperature and R the gas constant. Setting ΔG‡ to zero gives us a rate constant of ~1014 s-1 (the barrier must be zero, or very close to it).

Here is the statue atop the plinth above. Apparently it is honoured by the students with robes and other accoutrements on special occasions.
‡Another famous timescale inference was by Beckmann in 1889[cite]10.1002/jlac.18892500306[/cite] when he deduced the existence of a transient unseen intermediate in the (what he thought was) racemisation of menthone. That intermediate of course was the enol.