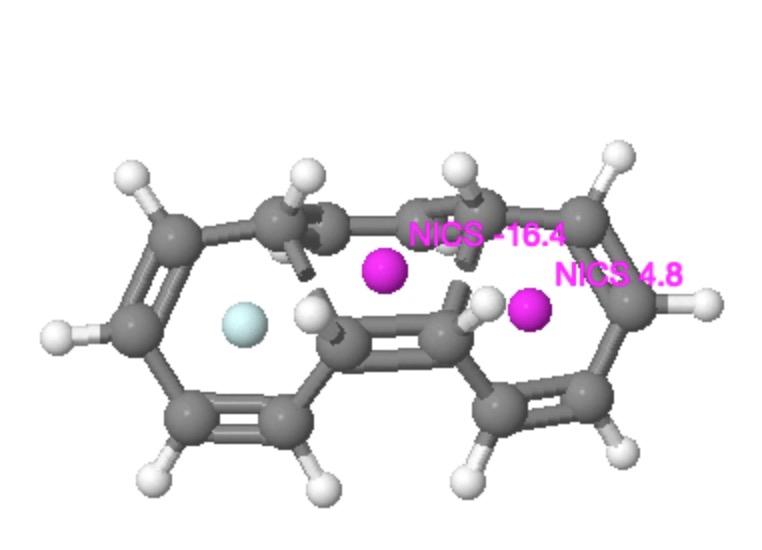
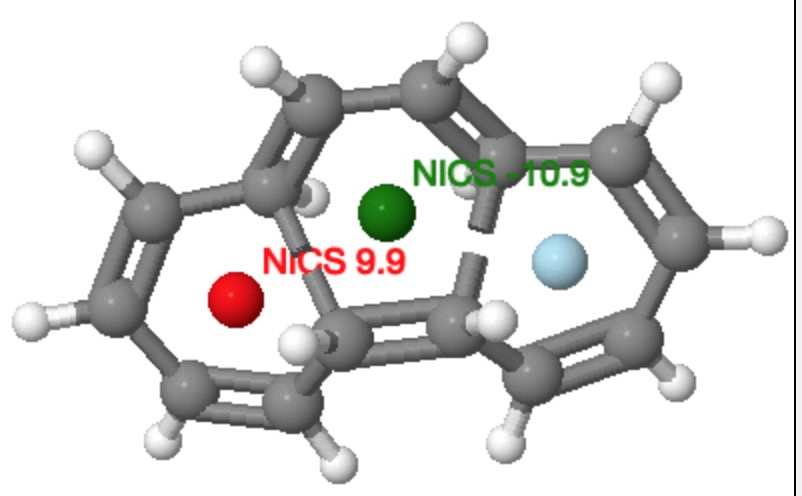
Much like climbing Mt. Everest because its there, some hypothetical molecules are just too tantalizing for chemists to resist attempting a synthesis. Thus in 1964, Edgar Heilbronner speculated on whether a conjugated annulene ring might be twistable into a Möbius strip. It was essentially a fun thing to try to do, rather than the effort being based on some anticipated (and useful) property it might have. If you read the original article (rumour has it the idea arose during a lunchtime conversation, and the manuscript was completed by the next day), you will notice one aspect of these molecules that is curious by its absence. There is no mention (10.1016/S0040-4039(01)89474-0) that such Möbius systems will be chiral. By their nature, they have only axes of symmetry, and no planes of symmetry, and such molecules therefore cannot be superimposed upon their mirror image; as is required of a chiral system (for a discussion of the origins and etymology of the term, see 10.1002/chir.20699).

- If the magnitude of the rotation is > 100°, then the sign of this rotation can be very reliably matched to either enantiomer. This allows the absolute configuration to be assigned with a lot of confidence, and probably much more easily than trying to do it by other methods.
- The magnitude itself can be reliably predicted to within 10% of the true value if the molecule is conformationally rigid. However, if it has any rotatable groups (and that even includes e.g. OH groups), then the result can be enormously sensitive to that conformation (or Boltzmann mixture of conformations). Put the other way, calculating the optical rotation could be regarded as a very sensitive way of determining conformations!
So what of the 16-annulene synthesized by Herges and co-workers. Well at the B3LYP/6-311G(2df,2pd) and SCRF(CPCM,solvent=chloroform) level of theory (which is reasonably accurate, although one can do better of course), the enantiomer shown by clicking on the graphic above is predicted to have a rotation of -1355° (for the digital repository entry for the calculation, see 10042/to-2176). That is indeed a large value for such a relatively small molecule, and is probably more reliable because of the lack of conformational ambiguity. Well, you saw the prediction here! Anyone up for testing it experimentally?
![A [24] annulene. Click on image for model.](http://www.ch.ic.ac.uk/rzepa/blog/wp-content/uploads/2009/04/gaytab.jpg "gaytab")














![Electrocylization of [14] annulene](http://www.ch.ic.ac.uk/rzepa/blog/wp-content/uploads/2009/04/p322.jpg "Electrocylization of [14] annulene")