Previously, I looked at models of how ammonia could be protonated by water to form ammonium hydroxide. The energetic outcome of my model matched the known equilbrium in water as favouring the unprotonated form (pKb ~4.75). I add here two amines for which R=Me3Si and R=CN. The idea is that the first will assist nitrogen protonation by stabilising the positive centre and the second will act in the opposite sense; an exploration if you like of how one might go about computationally designing a non-steric superbasic amine that becomes predominantly protonated when exposed to water (pKb <1)† and is thus more basic than hydroxide anion in this medium.

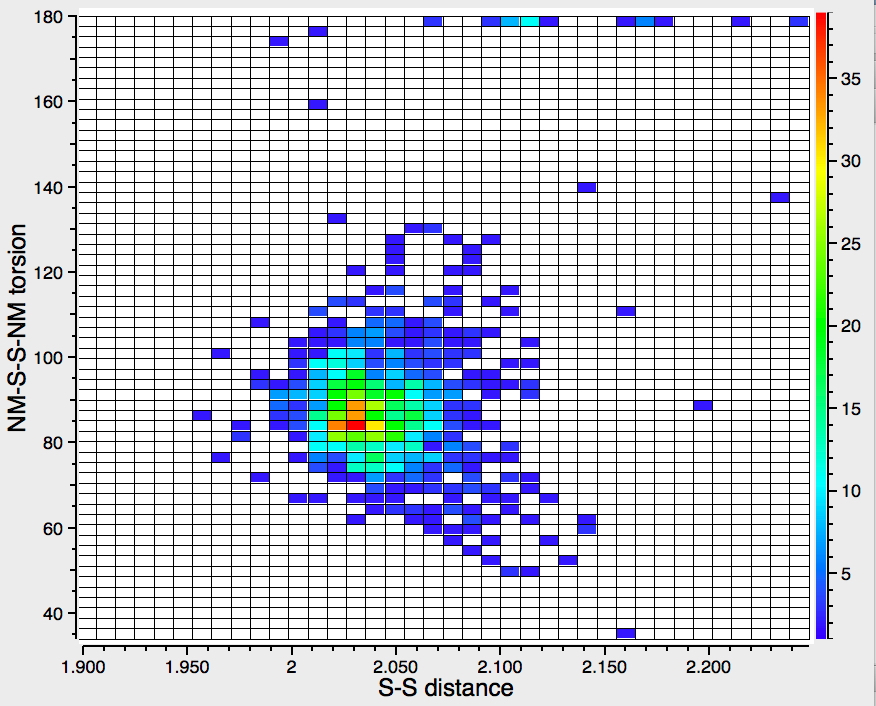
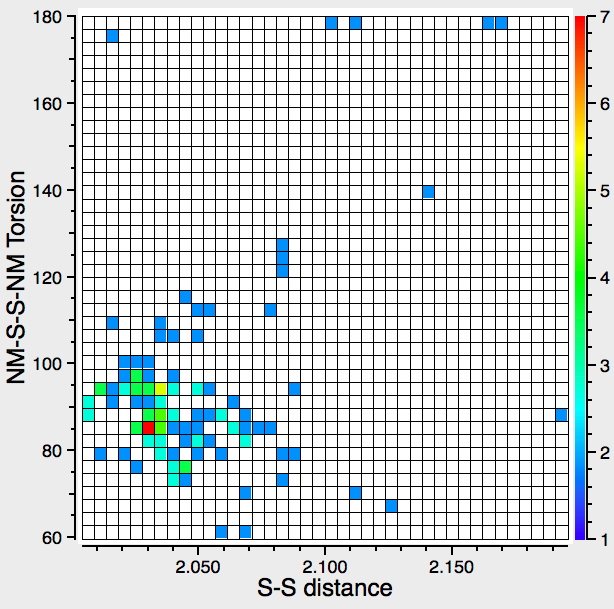
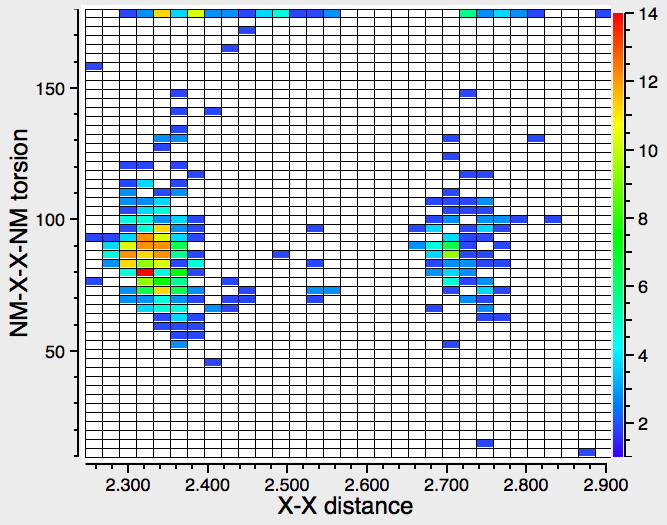
Before reporting any calculations, let us see what the CSD (Cambridge structure database) might contain. The search query is simple, a 3-coordinate amine forming a 4-coordinate quaternary nitrogen† with one N-H and a positive (formal) charge on the N, and a 1-coordinate oxygen with one O-H and a negative charge on the O. With the constraints R < 10%, no disorder and no errors, one gets as many as 15 hits,[cite]10.14469/hpc/361[/cite] several of which also apparently contain separate water molecules in the crystal. A warning bell (perhaps several) sounds, since if R < 5%, the number of hits drops to just 2; these are clearly difficult structures to refine!‡ So there is some tantalising evidence that in the solid state at least, the quaternary ammonium group (with at least one N-H), water and a hydroxide anion might be capable of co-existence. As noted below† some fascinating 2-coordinate amines have also been reported as having superbasic properties.
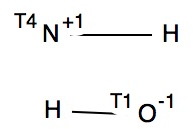
R=CN: the well known compound cyanamide is known to act only as an acid and its basic properties are never quoted. Shown below is the reaction path for transfer of a proton from water to the amine using an 8-water model (n=8) in which two bridges can serve to help stabilize any ionic form. The energy required to do so however is at least 24 kcal/mol (ωB97XD/Def2-TZVPPD/SCRF=water) which indicates that no protonated amine is formed. This can be attributed to the electron withdrawing cyano group strongly destablising any adjacent positive ammonium centre and thus effectively completely inhibiting its formation.

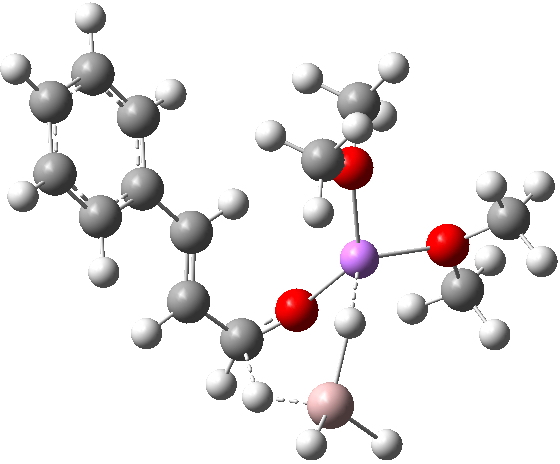
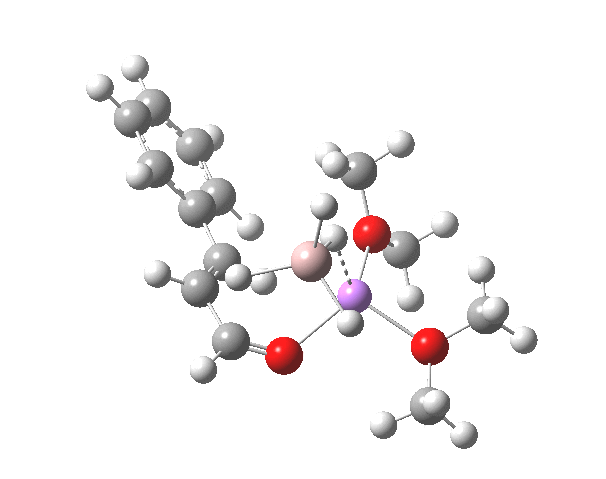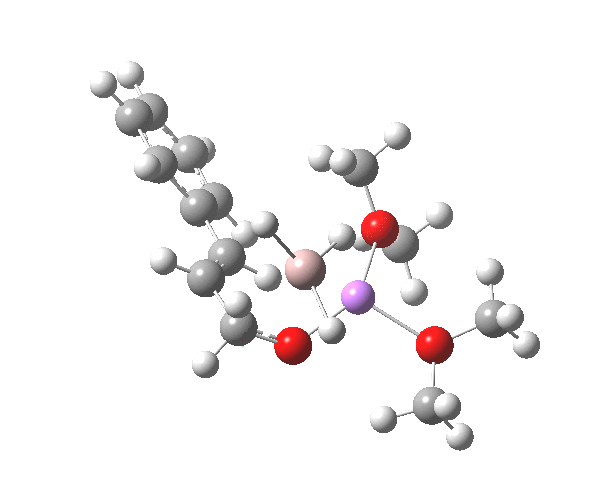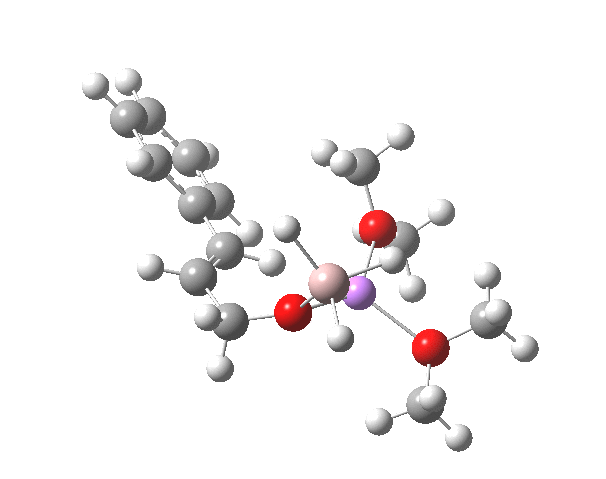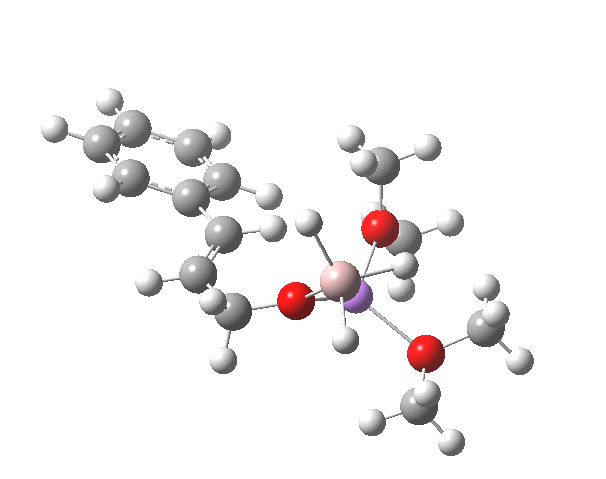
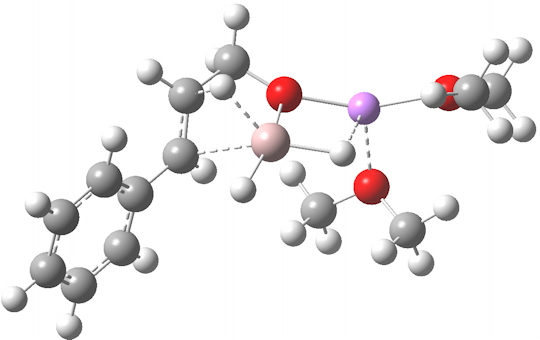
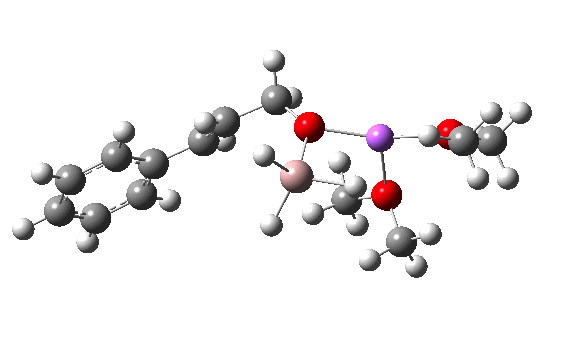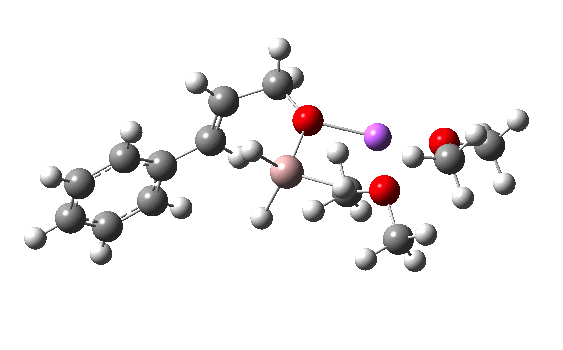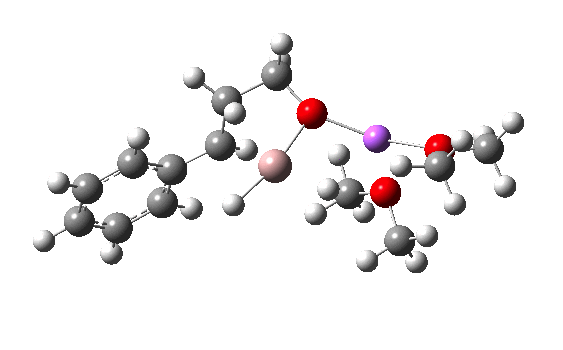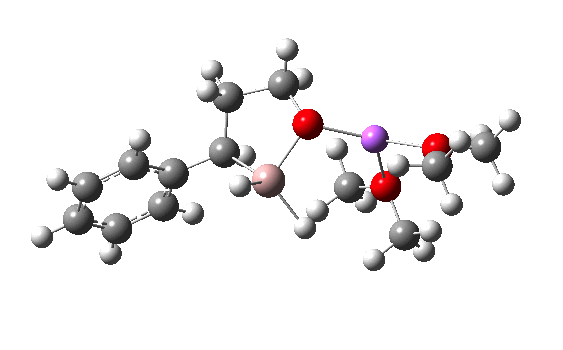
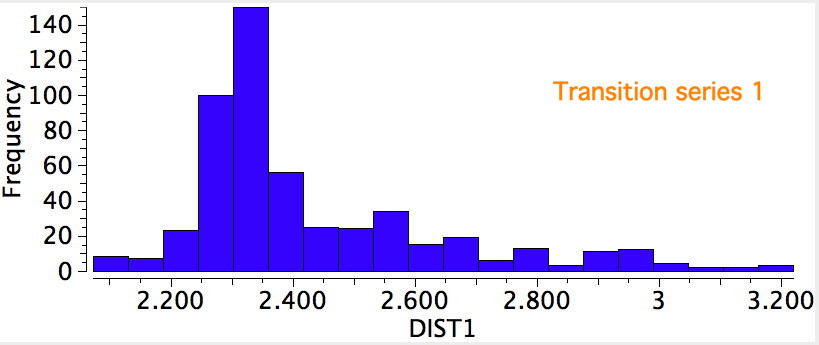
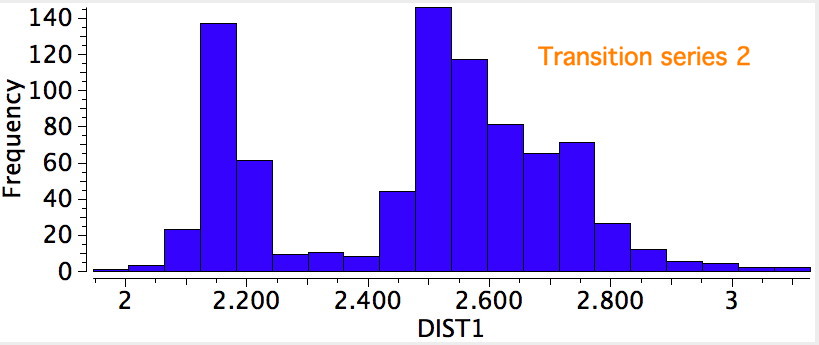
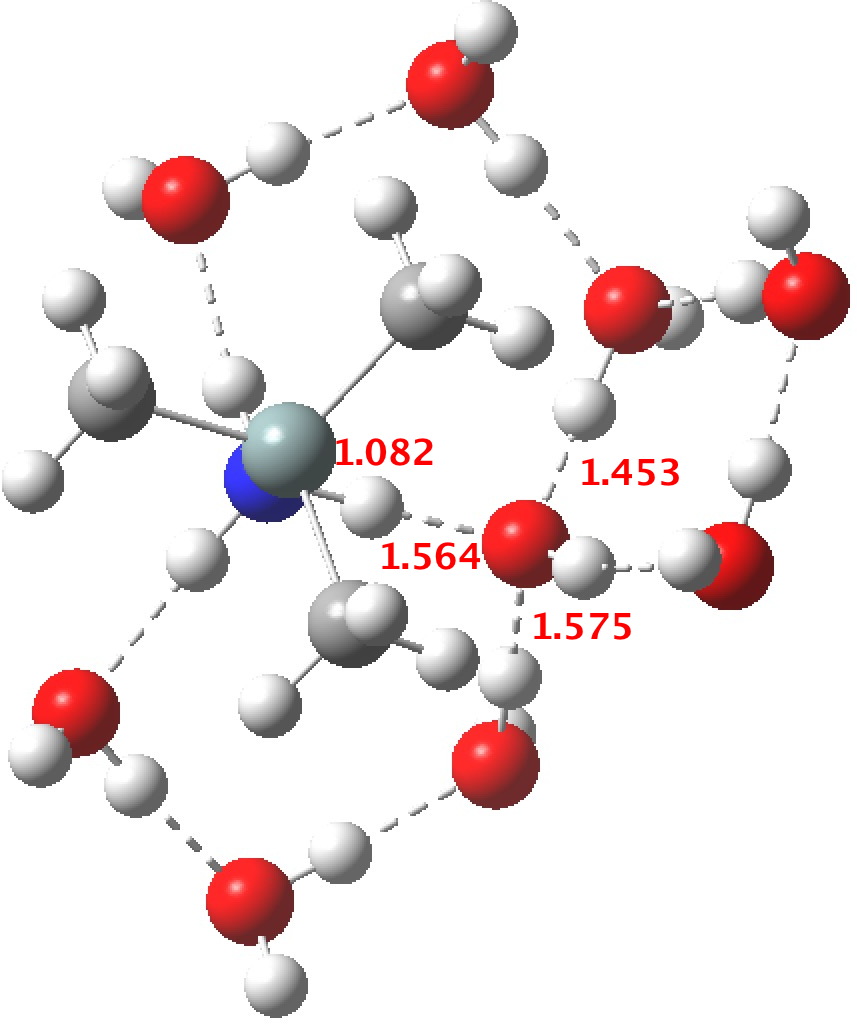
R=Me3Si: this too is already known[cite]10.1016/j.jorganchem.2006.09.021[/cite],[cite]10.5517/CCNDXZX[/cite] but only in the presence of the non-coordinating counter-anion B(C6F5)4 crystallised from non-protic solution. An ionised form can now be located using the model above. This has the structure shown below; note the very short hydrogen bonds associated with the hydroxide anion and the possibility of forming only two water bridges across the ion-pair. The relative free energy of the ion-pair (table below) shows it to be if anything less basic than ammonia.
| n=8 | R=H | R=SiMe3 | R=CN |
|---|---|---|---|
| ΔΔG298 | 7.0[cite]10.14469/ch/191946[/cite] |
7.6[cite]10.14469/ch/191987[/cite],[cite]10.14469/ch/191982[/cite] |
>24[cite]10.14469/ch/191983[/cite] |
NBO (natural bond orbital) analysis might here be a useful metric of basicity. Hence Me3SiNH2…H2O suggests that donation from the N lone pair into an antiperiplanar Si-C bond is quite large (E(2) = 11.9 kcal/mol), although alternative donation by nitrogen into the H-O σ* bond of the water is much higher (33.4 kcal/mol).
Perhaps the basicity of simple amines is related to their ability to form stabilizing water bridges across the ion-pair? With trimethylsilyl substituents, this feature (and hence the basicity) is partially or even fully suppressed as in e.g. tris(trimethylsilyl)amine.The pKb of the latter appears to be unreported[cite]10.1039/tf9646001257[/cite] but it does seem to be only weakly basic and "inert to H2O",[cite]10.1002/zaac.19603030502[/cite] a property attributed instead to multiple character in the Si-N bonds.
I will in a future post look at the alternative class of phosphazenium amines which do manage to achieve superbasicity.[cite]10.5517/CC12MRFW[/cite]
†A phosphazenium 3-coordinate amine[cite]10.1002/anie.198711671[/cite] was in 1991 claimed to be the strongest metal-free neutral base. This has now been superceded by combining this base motif with that of a sterically operating proton sponge.[cite]10.1021/ja409760z[/cite],[cite]10.5517/CC12MRFW[/cite] I will report the computational modelling of these systems in a later post.
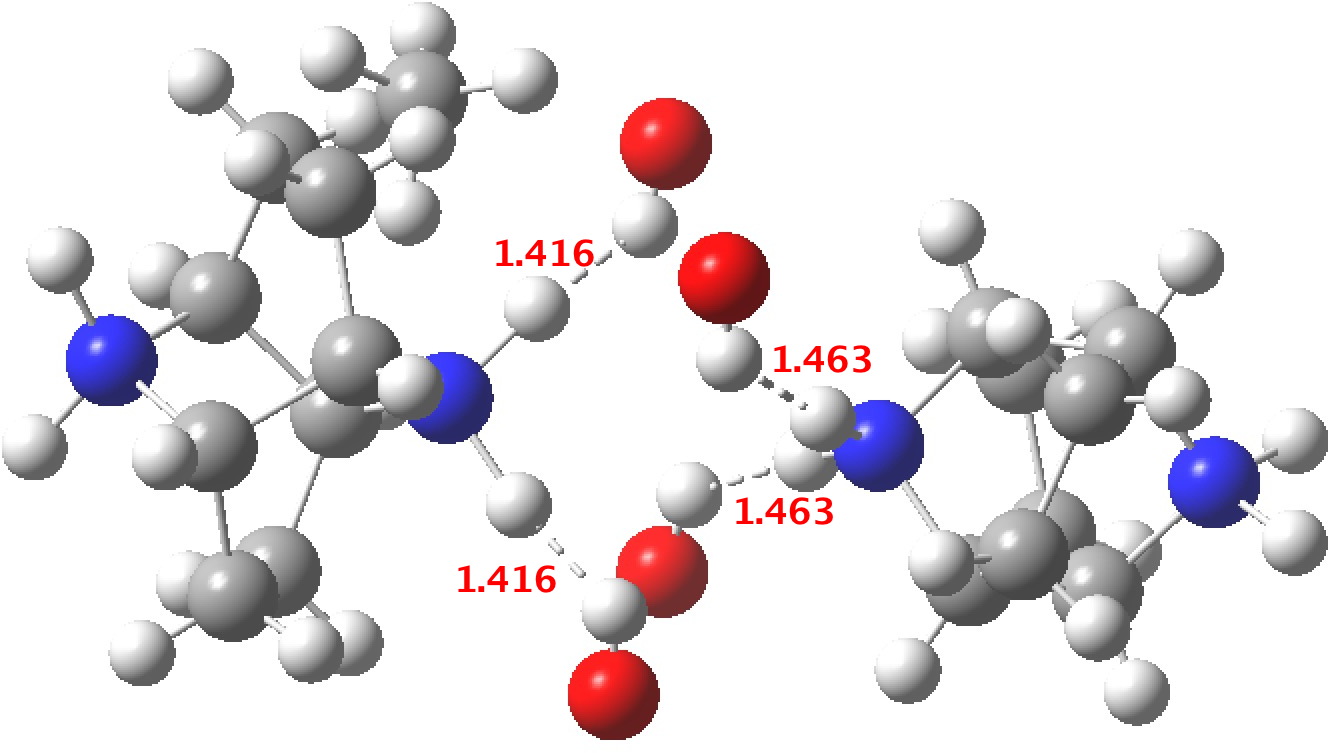
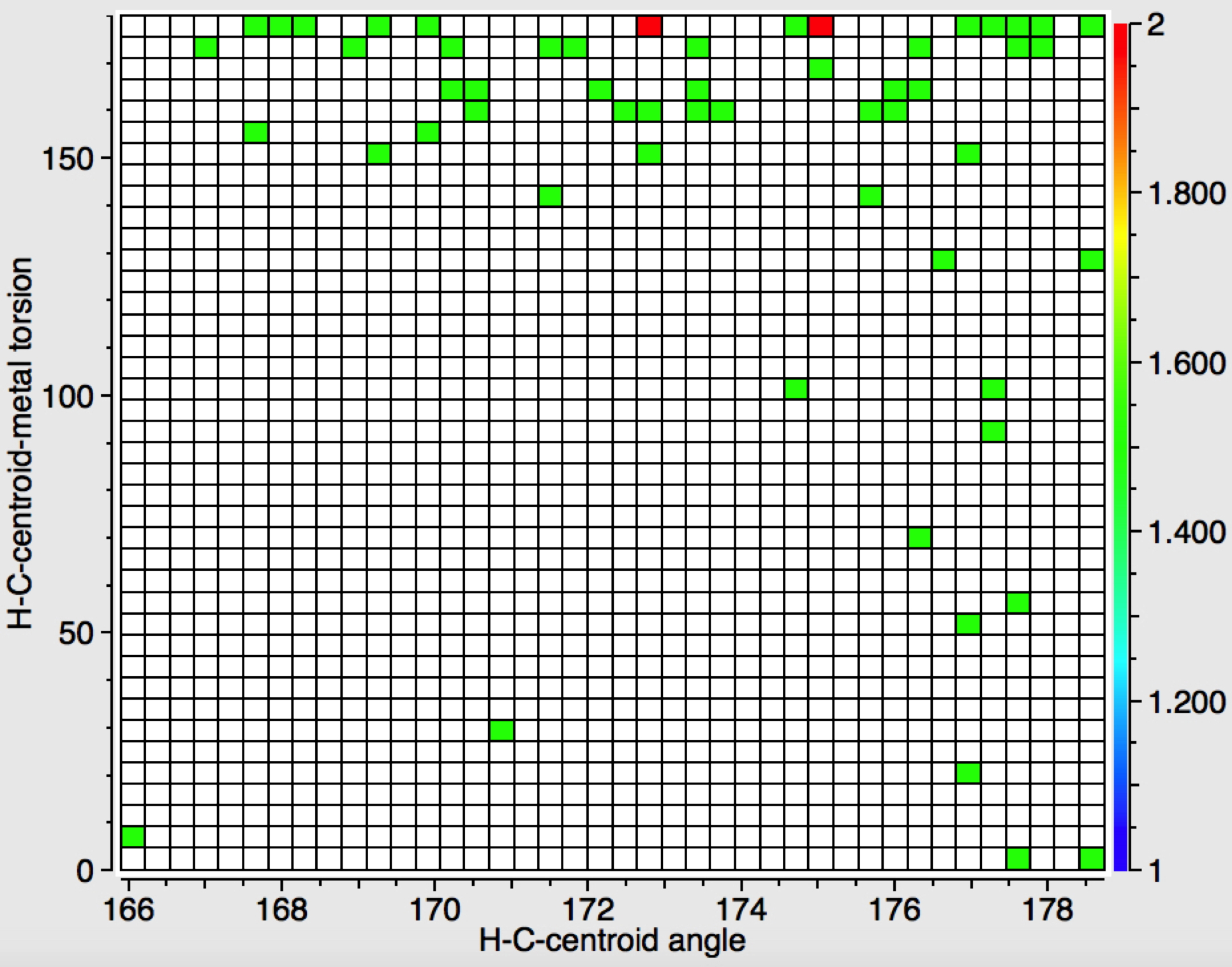
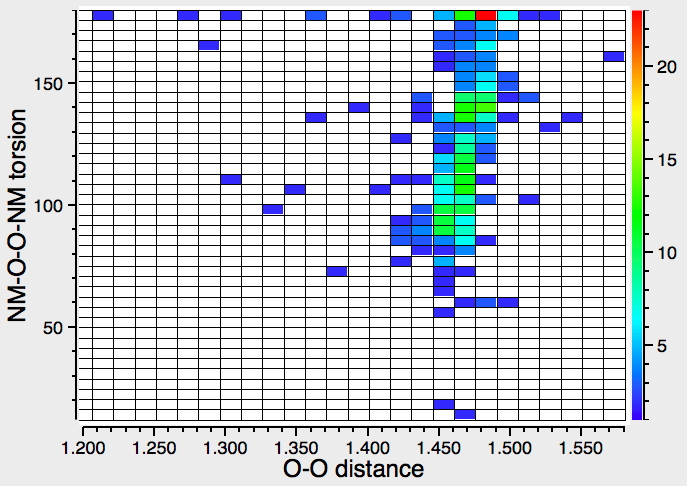
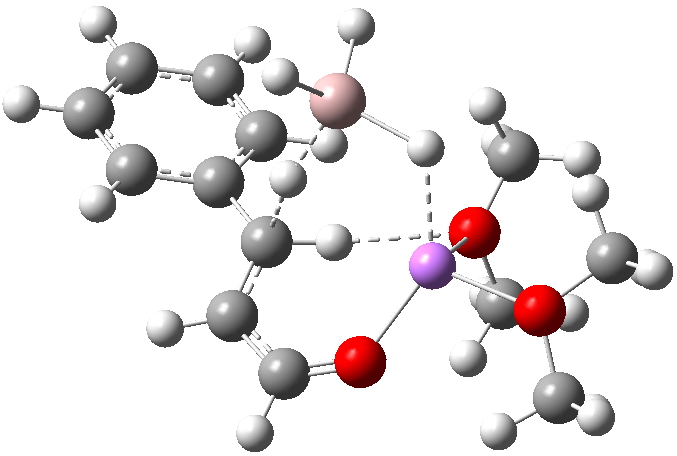
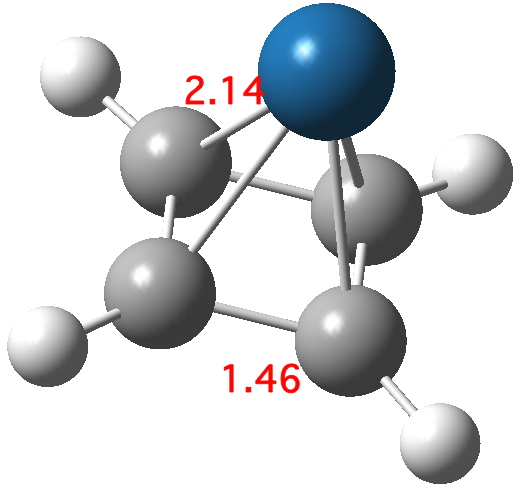
‡One of the structures identified with R<10% is UBEJIU[cite]10.1021/jm991140q[/cite] and which is worth showing below. Note the apparent close contact of the type N-H…H-O (1.416-1.463Å) rather than the expected N-H…OH. If correct (this feature is not mentioned in the article itself) it would be classified as a dihydrogen bond, a type normally only found in situations such as B-H…H-N. There are a number of other inconsistencies which must be resolved if this structure is to stand as correct.