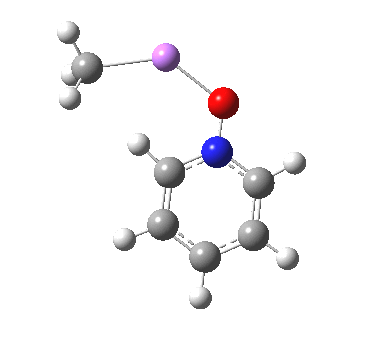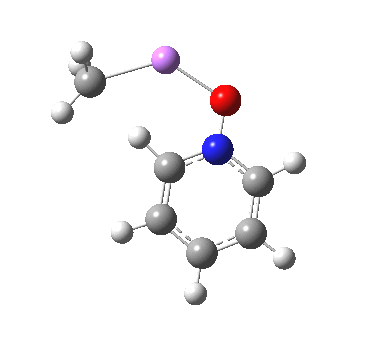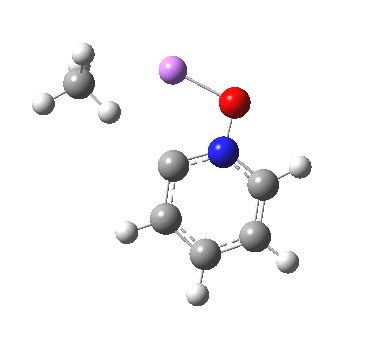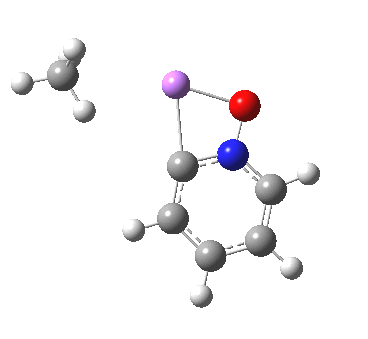
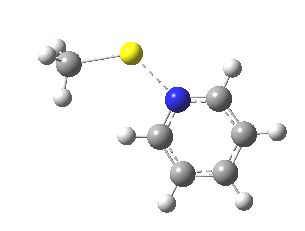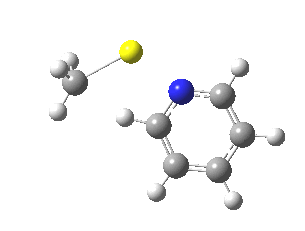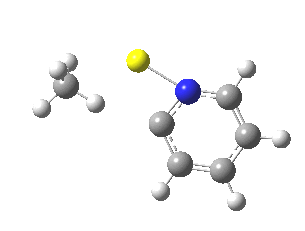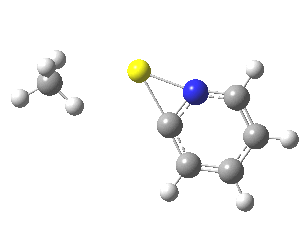
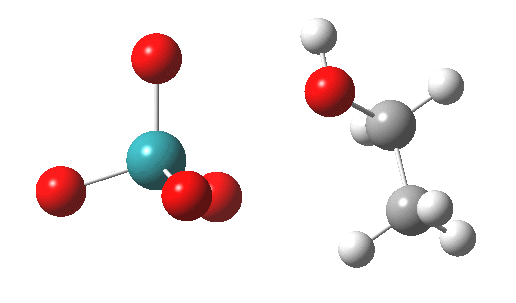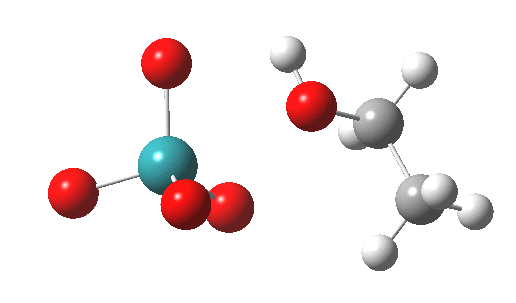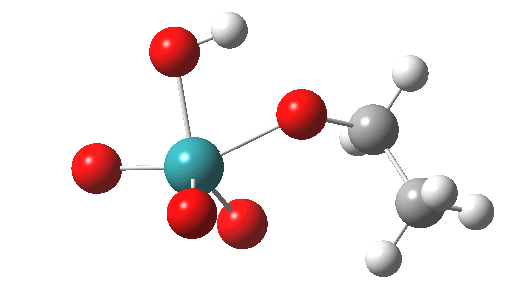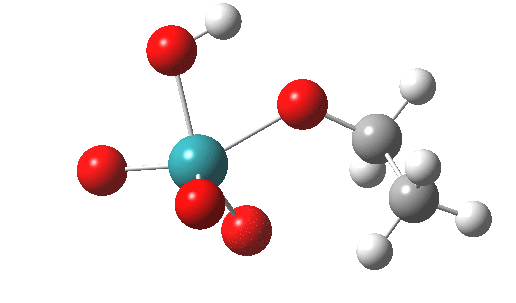
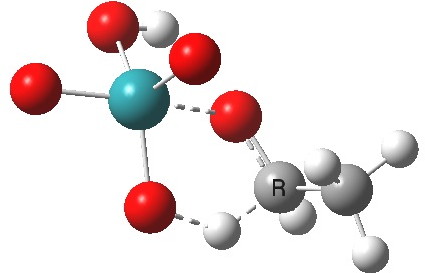
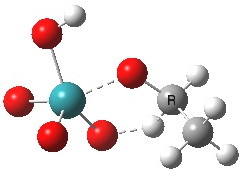
This is another in the occasional series of “what a neat molecule”. In this case, more of a “what a neat idea”. The s-triazine below, when coordinated to eg ZnI2, forms what is called a metal-organic-framework, or MOF. A recent article[cite]10.1038/nature11990[/cite] shows how such frameworks can be used to help solve a long-standing problem in structure determination, how to get a crystal structure on a compound that does not crystallise on its own.

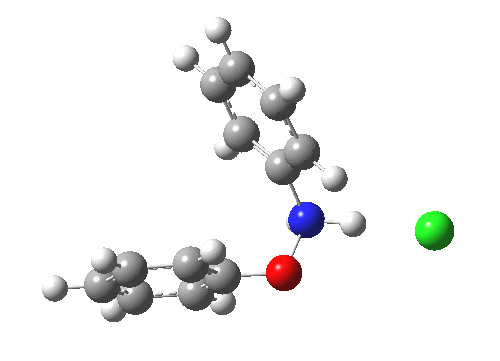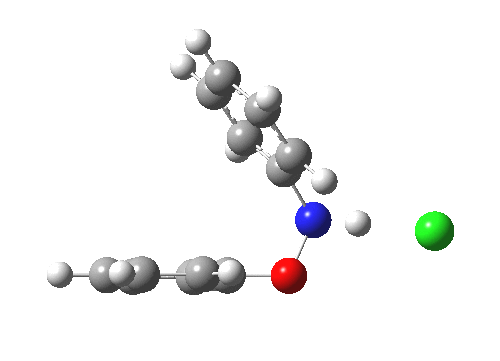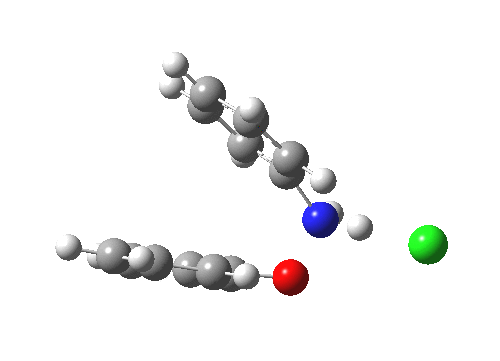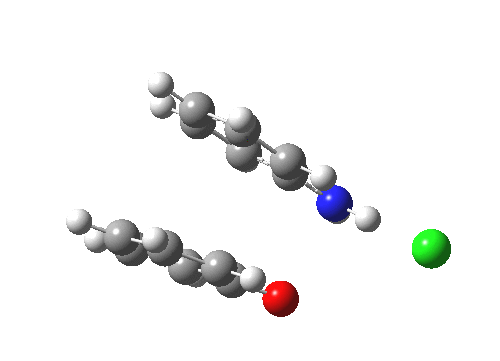
The essence of the technique is to select a small crystal of the MOF (which crystallises well) and allow your own molecule to diffuse in from solution. So captured inside the framework, the X-ray analysis can now be done on the absorbed host molecule together with the MOF framework. Below you can see two of the structures reported solved by this technique. The first shows the target molecule (green arrows) but also three molecules of cyclohexane (the diffusing solvent, red arrows), nicely illustrating its chair conformation.
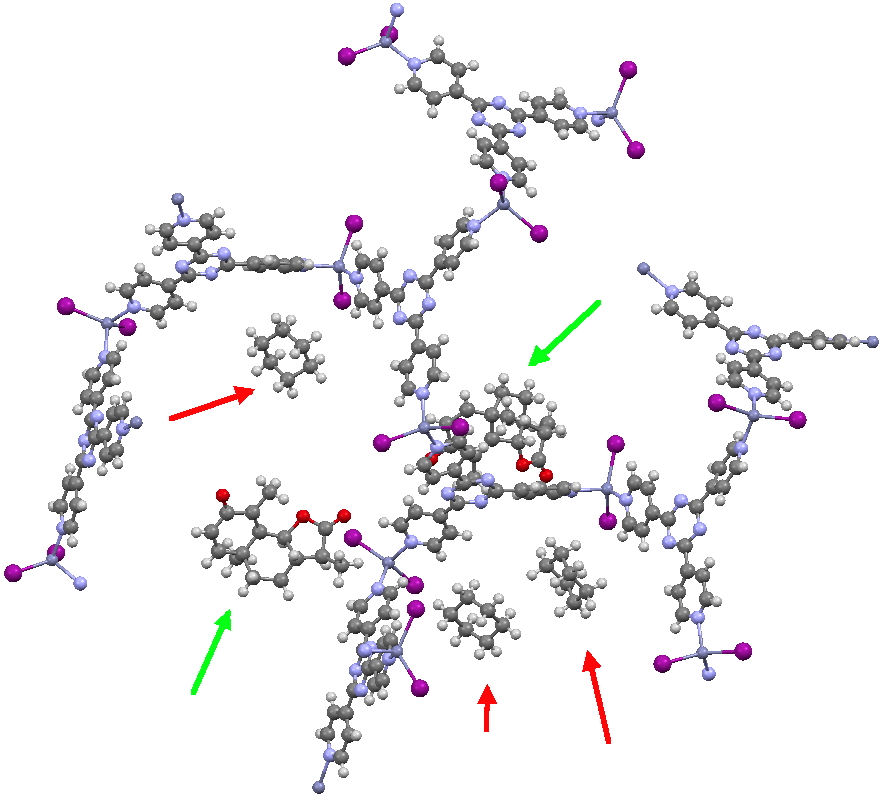
This second example shows the structure of a marine natural product, of which only about 5µg was available (green arrow). The structure (of miyakosyne A) shows a conformationally flexible substituted saturated backbone, a molecule which traditionally might be expected to be disordered because of its flexibility. These structures were performed on a standard diffractometer, and the authors point out that if more intense synchrotron radiation were to be used, even smaller samples (< 10ng) could be determined. They also note that full occupancy of the MOF lattice does not need to be achieved for the method to succeed.
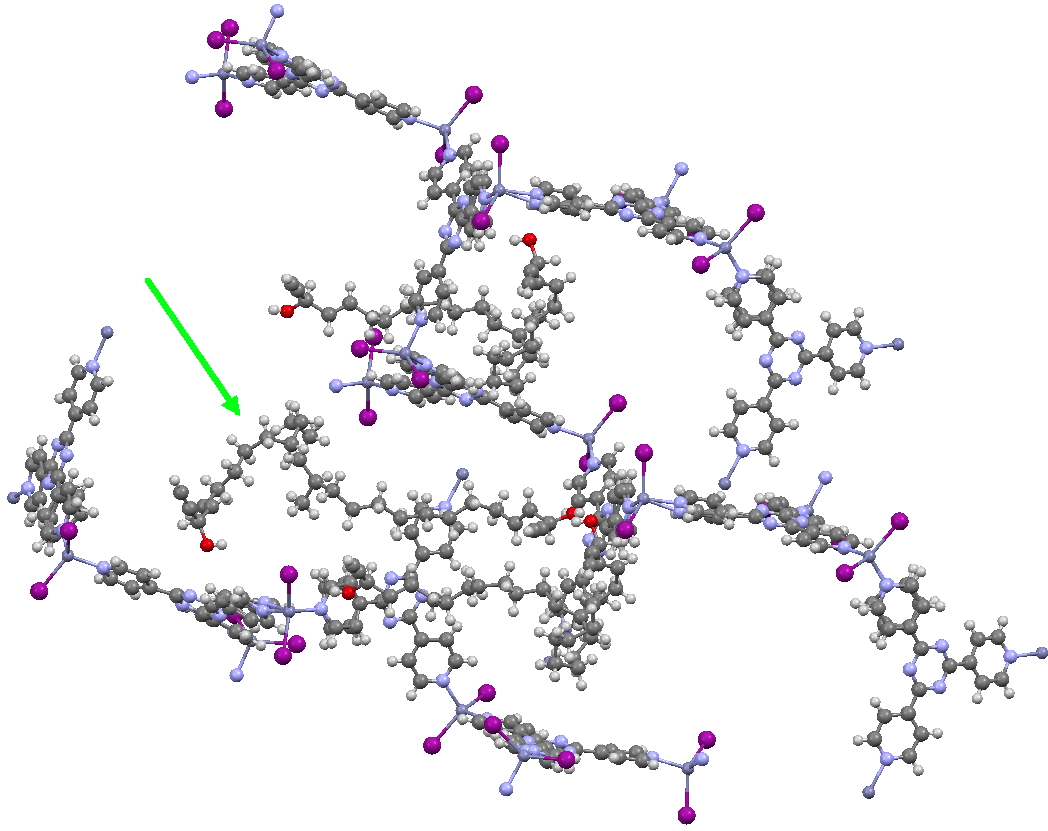
I end with noting that in an earlier post, I observed how chiroptical methods can nowadays be increasingly successfully used to determine absolute configurations of chiral molecules. Miyakosyne A, a molecule with three chiral centres had proved to be a challenge using such techniques; one of these centres (at C14) could not be determined by any chiroptical method. The configuration was however successfully determined as (S) by this new crystallographic technique. I think this is a huge contribution to the science of structure and configuration determination![cite]10.1002/(SICI)1521-3773(19990115)38:1/2<153::AID-ANIE153>3.0.CO;2-U[/cite]