Science frequently works by people making connections between related (or even apparently unrelated) concepts or data. There are many ways of helping people make these connections – attending a conference or seminar, searching journals for published articles and nowadays also searching for data are just a few examples. For about 20 years now, one technology which has been helping to enable such discoveries is what are called “Persistent IDentifiers” or PIDs. These are unique labels which can be attached to a (scientific) object such as a journal article, a dataset or a researcher. The PIDs for the first two examples have become better known as DOIs (digital object identifier), the last is known as an ORCID. The PID is registered with a registration authority. Two of the oldest and best known authorities are CrossRef for journal articles, funders (etc) and DataCite, who specialise in citable identifiers for data. The registration process includes creating and adding a metadata record to the PID, the record is then indexed and can then be used for searching for the objects. The terms of these metadata records are carefully controlled to use specified and standardised vocabularies to describe the objects (one current initiative in chemistry in this area is described here[cite]10.1515/pac-2021-2009[/cite]).
The PID “ecosystem” is constantly expanding and a recent addition is the ROR registration authority. This issues PIDs for research organisations, so that one can then easily associate a scientific object with the organisation where the research was conducted. The initial focus for ROR PIDs was the traditional forms of organisation such as a university and company research labs. Here I tell about how a rather different type of organisation came to have its own ROR, the “World Association of Theoretical and Computational Chemists” or WATOC. The aims of WATOC are primarily to hold triennial congresses to promote scientific exchange and to help researchers make those connections through presentations, posters and numerous coffee breaks!
Last July, the proposal for creating a ROR for WATOC was accepted by its decision making body and can now be announced as https://ror.org/04rp40h82, where 04rp40h82 is the unique WATOC identifier. The prefix https://ror.org/ is called the “resolver”, which in turn allows access to the associated metadata record via an API. That record in turn includes a link to the organisation, similar to links to journal articles as specified by a DOI.
It is now time to show some examples of how the WATOC ROR can actually be used.
- One outcome of the last WATOC Congress held in 2022 in Vancouver is the production of a themed peer-reviewed issue of the Canadian journal of chemistry, created by inviting speakers to submit an article corresponding to their presentation. Armed with the WATOC ROR, the publisher was approached to ask if this identifier could be included in the metadata record for each accepted article. This was agreed and in due course will be added to the Crossref metadata record for each article in this special issue. When this happens, it can be searched using e.g. https://api.crossref.org/works?filter=ror-id:04rp40h82 Because creation of a metadata record is actually part of the complex journal production workflow, this will not occur until the journal has updated its procedures to do this, which may take a little while yet. Invoking that search would then allow all published articles associated with (at least in part) WATOC activities.
- The link https://api.crossref.org/works?filter=ror-id:04rp40h82 is actually part of the CrossRef API (application programmer interface) and so can now be used to construct complex programatic queries which include the WATOC ROR and for deployment in e.g. AI applications.[cite]10.1145/3447772[/cite] Although not derived from the CrossRef API, I can show here some similar uses of metadata for the construction of so-called Knowledge Graphs [cite]10.1145/3447772[/cite], which can be thought of as visual representation of connections between scientific objects, organisations and other types of entity to which a registered PID has been assigned.
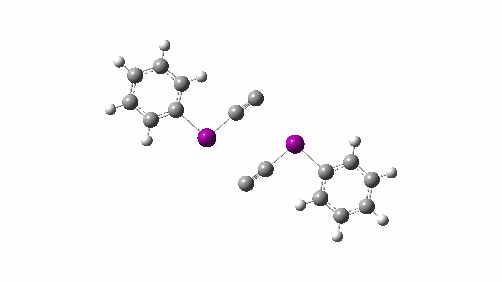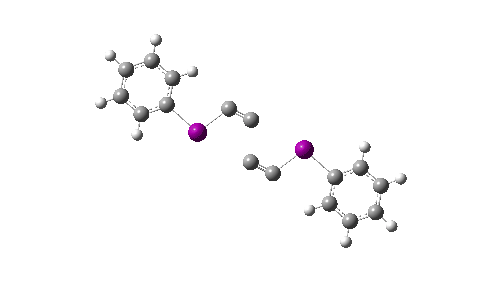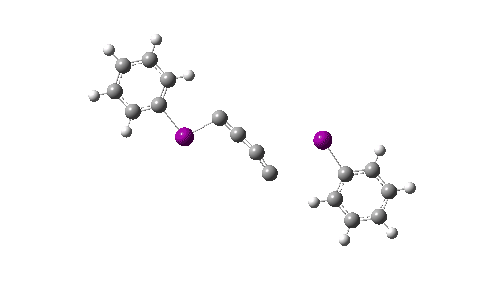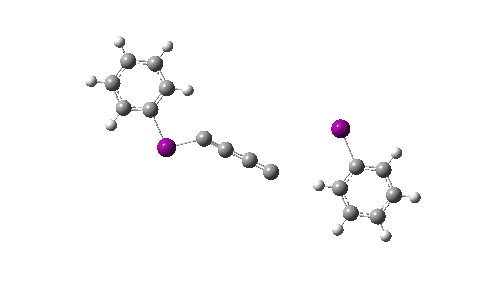
- This knowledge graph was created using SciFinder by specifying a person (myself in this case) and any conferences they have been associated with. However, in the past the capture of conference attendance was a rather hit and miss process and so the record is very incomplete. It is the expectation that metadata associated with ROR PIDs will help make these records more complete and hence useful. ROR is also fully open and hence its use is less restricted than the proprietary SciFinder system.
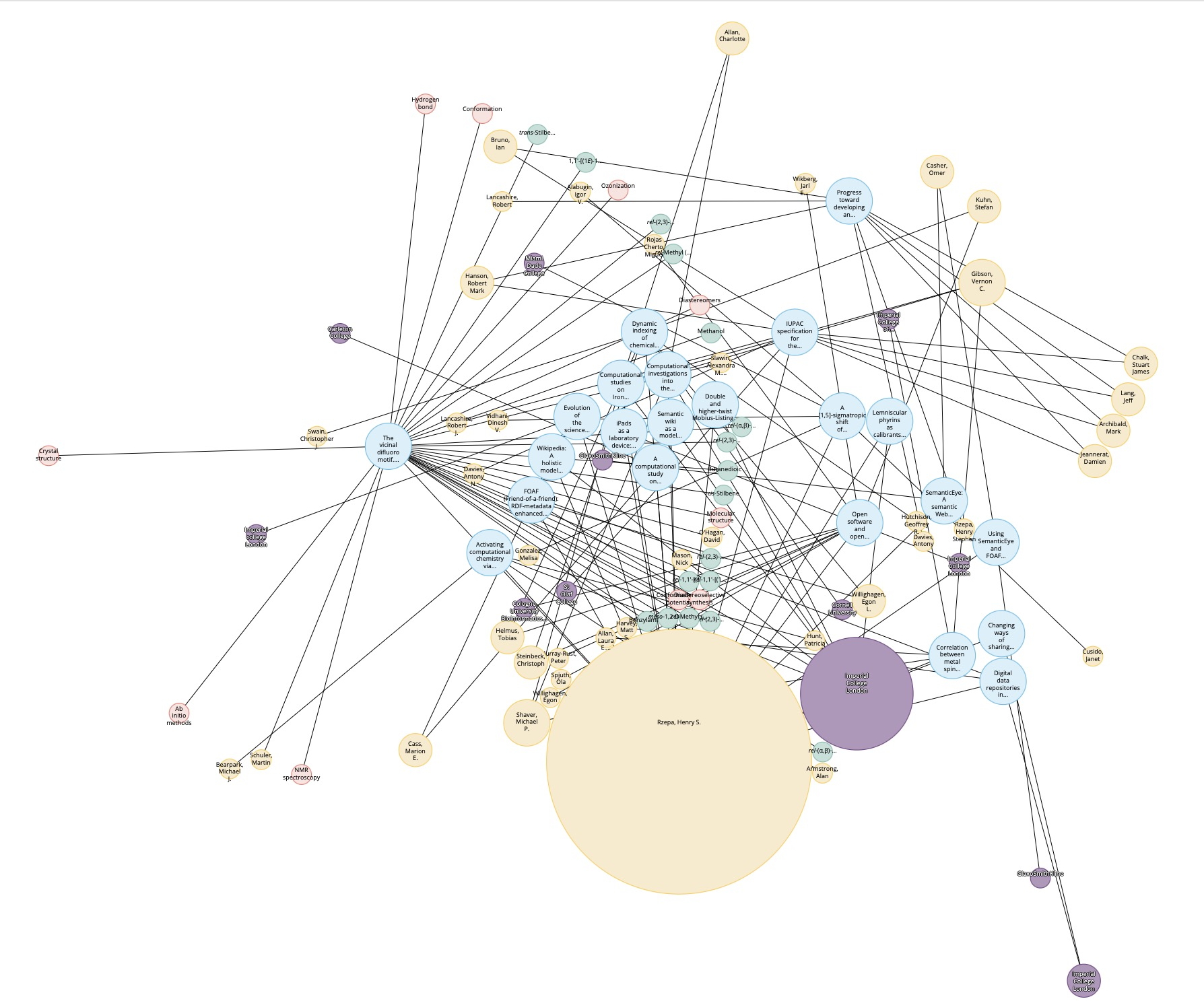
- I cannot resist also adding this one. The metadata record now contains named concepts, this one being “transition states” which I have been associated with in the past.

- As of today, the WATOC ROR has not propagated to any CrossRef metadata records and so I cannot yet show any knowledge graphs with nodes based on WATOC.
- This knowledge graph was created using SciFinder by specifying a person (myself in this case) and any conferences they have been associated with. However, in the past the capture of conference attendance was a rather hit and miss process and so the record is very incomplete. It is the expectation that metadata associated with ROR PIDs will help make these records more complete and hence useful. ROR is also fully open and hence its use is less restricted than the proprietary SciFinder system.
-
The ROR PID can also be used for inclusion in metadata records describing datasets. This is one such search, now of the DataCite metadata store:
https://commons.datacite.org/doi.org?query=((contributors.affiliation.affiliationIdentifier:*04rp40h82)+AND+(contributors.affiliation.affiliationIdentifierScheme:ROR))+OR+((creators.affiliation.affiliationIdentifier:*04rp40h82)+AND+(creators.affiliation.affiliationIdentifierScheme:ROR))
Note the somewhat more complex logic being used, in part because a dataset can be “created” by a named person but also can be “contributed to” and one should really search for both possibilities. -
One can also combine two different identifiers, namely an organisational ROR and a researcher ORCID into a single query:
https://commons.datacite.org/?query=((creators.affiliation.affiliationIdentifier:*04rp40h82)+OR+(contributors.affiliation.affiliationIdentifier:*04rp40h82))+AND+(contributors.nameIdentifiers.nameIdentifier:*0000-0002-8635-8390)
There are many more combinations of searches that can be constructed using other types of identifiers.[cite]10.1002/mrc.5186[/cite] - Further in the future, one might expect that metadata records from e.g. both CrossRef and DataCite could be combined to create knowledge graphs by combining information based on both journal articles and published FAIR datasets. Currently, CrossRef does not identify PIDs for datasets that might be cited in an article bibliography as explicit data, but that too may be coming in the near future.[cite]10.3789/niso-rp-36-2020[/cite]
Way back in January 1994, WATOC was one of the very first chemical-science based organisations to have its own web page. Now it is leading the way in acquiring and deploying its very own persistent identifier in the form of a ROR. One might hope that many more such organisations acquire one soon.
The DOI for this post is 10.14469/hpc/12363