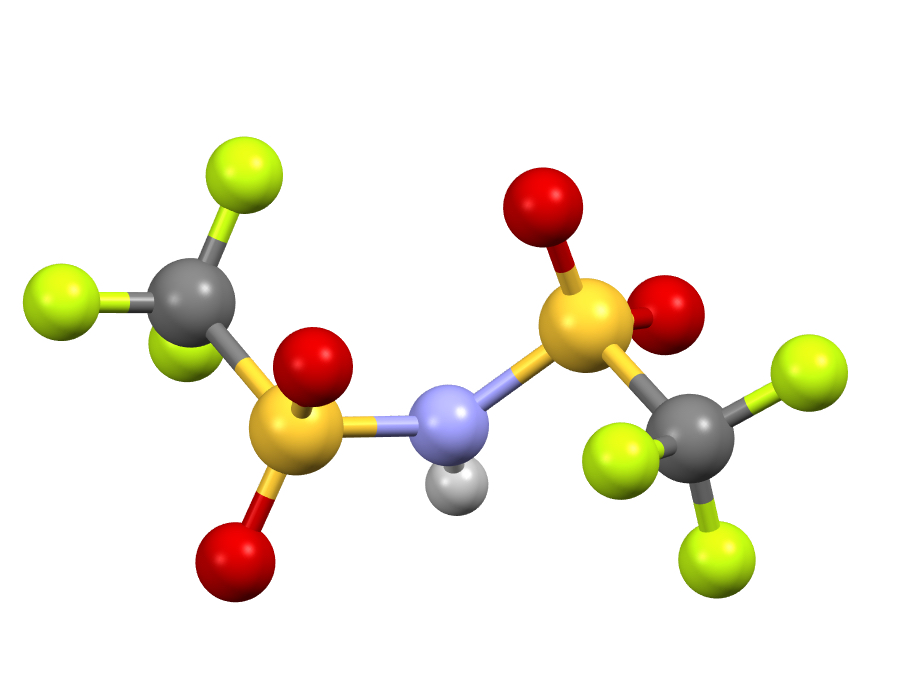
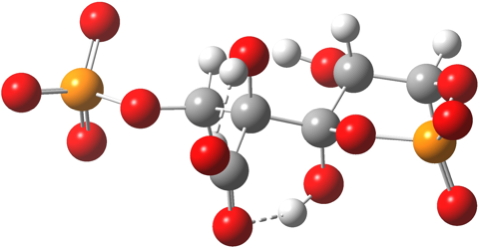
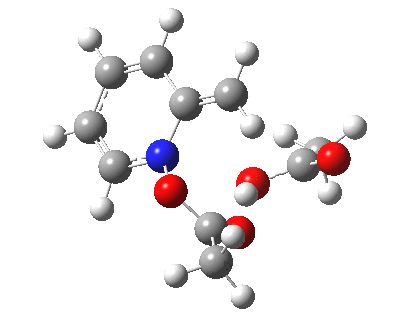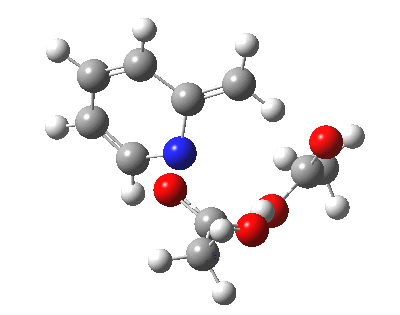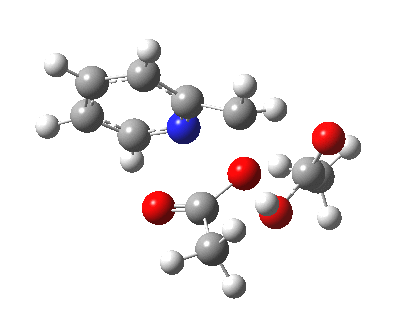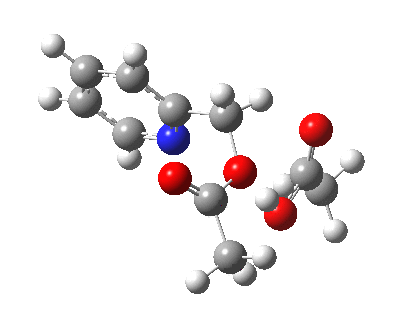
Steganone is an unusual natural product, known for about 40 years now. The assignment of its absolute configurations makes for an interesting, on occasion rather confusing, and perhaps not entirely atypical story. I will start with the modern accepted stereochemical structure of this molecule, which comes in the form of two separately isolable atropisomers.

The first reported synthesis of this system in 1977 was racemic, and no stereochemistry is shown in the article (structure 2).[cite]10.1039/P19770001674[/cite] Three years later an “Asymmetric total synthesis of (-)steganone and revision of its absolute configuration” shows how the then accepted configuration (structure 1 in this article) needs to be revised to the enantiomer shown as structure 12 in the article[cite]10.1016/S0040-4039(00)78586-8[/cite] and matching the above representation. The system has continued to attract interest ever since[cite]10.1039/P19820000521[/cite],[cite]10.1039/A900743A[/cite],[cite]10.1039/C39950001943[/cite],[cite]10.1002/ejoc.201402761[/cite], not least because of the presence of axial chirality in the form of atropisomerism. Thus early on it was shown that the alternative atropisomer, the (aS,R,R) configuration initially emerges out of several syntheses, and has to be converted to the (aR,R,R) configuration by heating[cite]10.1039/P19820000521[/cite]. One could easily be fooled by such isomerism!
Absolute configurations can be established in several ways.
- From precursors of known absolute configuration. This was the most common method until relatively recently, but it is very expensive since asymmetric syntheses are often much more complex and longer than racemic ones. There is always a small residual doubt that any transformation in the synthesis might have altered the configuration in an unexpected manner.
- From an X-ray of the final configuration (Bijvoet). Very often the structure is determined on a derivative of the target compound (the original may not form suitable crystals). There is also the doubt that the selected crystals may in fact be a minor form and do not represent the bulk of the system in solution. This is especially true where atropisomerism is concerned, since the solid state structure may not represent the same atropisomer present in solution.
- In the last decade or so, it has become more common to make use of the computation of measured chiroptical spectroscopies to see if they match. It turns out that this method appears never to have been applied to Steganone, and here I attempt to rectify this.
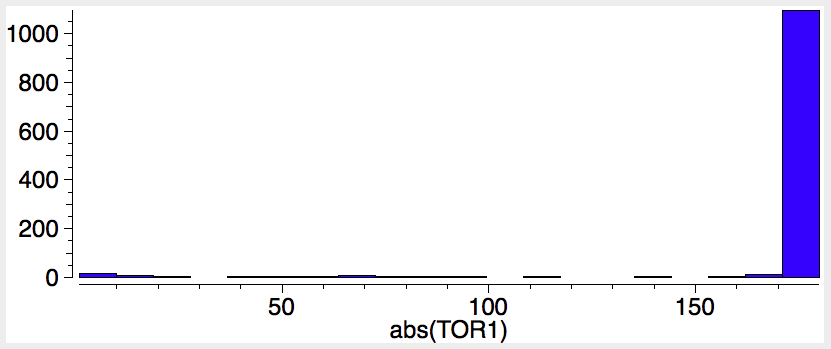
First, let us compute the optical rotation. The (aR,R,R) stereoisomer is also known as (-)-Steganone, because the measured specific rotation is [α]589 -170° ± 30.[cite]10.1039/P19820000521[/cite] It is computed (MN12L/6-311++G(d,p)/SCRF=chloroform) as -240°, [α]365 -2251[cite]10.14469/ch/189647[/cite]. The other atropisomer (aS,R,R) is computed to be 4.5 kcal/mol higher in free energy with [α]589 +408°[cite]10.14469/ch/189646[/cite], and measured as +150.[cite]10.1039/P19820000521[/cite] There is some uncertainty in the computed values, since the rotations can be dependent on the conformation not only of the rings, but the substituents. You might imagine that the conformation of eg a -OMe group is unimportant, but this is not so. In this case, I have used a crystal structure of a related species to serve as the start point for optimising the MeO conformations. The greater mismatch between computation and experiment for the (aS,R,R) stereoisomer probably needs an exploration of more conformations of the -OMe groups. At least in both cases the signs match between computation and measurement.
Next, the electronic circular dichroism (ECD), which has also been measured[cite]10.1039/P19820000521[/cite] for the (aR,R,R) isomer as Δε 201nm (-ve Cotton effect), 218 (+ve), 244 (-ve), 276 (+ve) 304 (-ve) and 337 (-ve). Bearing in mind that the baselines in ECD spectra are notoriously difficult to define (moving it up or down can easily invert a Cotton effect), the agreement with the calculated spectrum MN12L/6-311++G(d,p)/SCRF=chloroform, nstates=200)[cite]10.14469/ch/189649[/cite] might seem reasonable, although the calculated version has more peaks in the region 225-265 than are reported (e.g. 235, +ve, 265, -ve).

The (aS,R,R) isomer seems a less good fit. The +ve peak at 218 is missing, the +ve 276 peak matches better than the other isomer, but the 337nm peak is again the wrong sign.

Of course, in such a game it may be the DFT functional used for the simulation that itself might be misleading, MN12L in this case. Just to check, I also include the results using M062X[cite]10.14469/ch/189657[/cite] to see how variable these simulations might be. The measured peaks at 201, 218, 244 and 337nm match, but the ones at 276 and 304nm do not.

Although matching computed with measured ECD spectra is commonly used to assign absolute configurations of molecules, you can see from these results that the technique is not a cast iron one! Even scanning through myriad DFT procedures to find the one that fits best is probably not a complete solution either. Can anything be done to further increase confidence?
How about Vibrational Circular Dichroism (VCD) predictions?[cite][/cite],[cite]10.14469/ch/189651[/cite]. Like ECD, VCD is also sensitive to conformation, which is why some modern instruments have low temperature probes operating at close to 0K which strive to capture only a single lowest energy conformation (although of course in any simulation, you have to identify that conformation reliably!). At some stage in the future, the VCD spectra of steganone might indeed be measured, and hence compared with the below. It might serve to increase confidence in the chiroptical methods as a means of assigning configuration.


We might conclude from this short exploration of chiroptical spectroscopy that no one single measured or computed value can be absolutely definitive; rather it is the accumulation from various sources that builds up the case for a particular configuration. But at least the above simulations do serve to add some useful additional data for the record.