I asked a while back whether blogs could be considered a serious form of scholarly scientific communication (and so has Peter Murray-Rust more recently). A case for doing so might be my post of about a year ago, addressing why borane reduces a carboxylic acid, but not its ester, where I suggested a possible mechanism. Well, colleagues have raised some interesting questions, both on the blog itself and more silently by email to me. As a result, I have tried to address some of these questions, and accordingly my original scheme needs some revision! This sort of iterative process of getting to the truth with the help of the community (a kind of crowd-sourced chemistry) is where I feel blogs do have a genuine role to play.

TS1 in this scheme is modified from before to include an extra borane coordinating to the oxygen of the O-R group. I will include here the intrinsic reaction coordinate [computed at ωB97XD/6-311G(d,p)], since it shows some fascinating features.

One notes that the barrier for extrusion (R=H) is lower than before, due to the effect of the extra coordinated BH3 group. But notice the “bump” at an IRC value of ~4.0. If one inspects the gradients along the IRC, they reveal that the ejecting H-H molecule is tempted to coordinate to the boron to form a 5-coordinate species (a “hidden intermediate”) before abruptly changing direction and flying off into space!

You can see an animation by invoking this link or below:
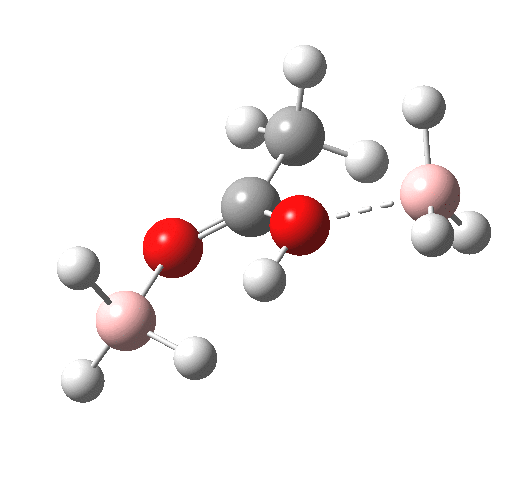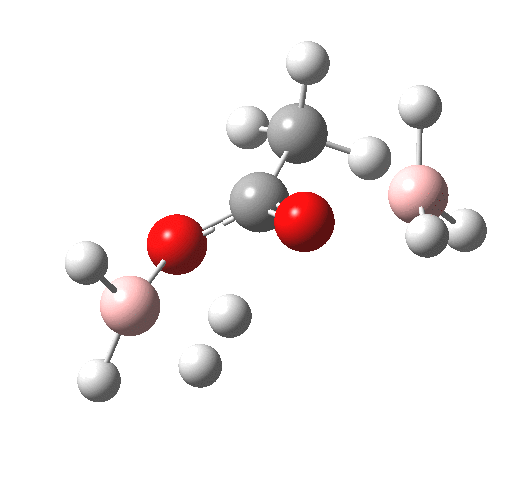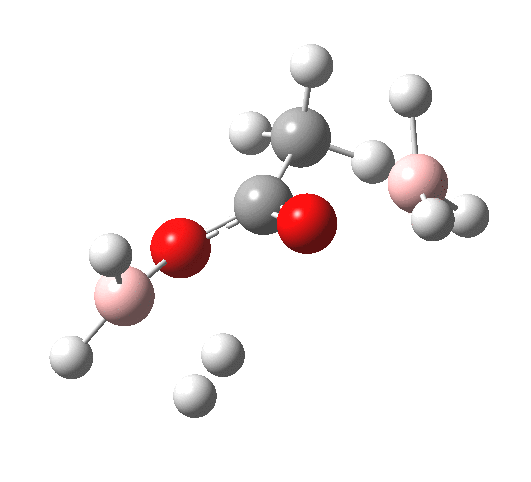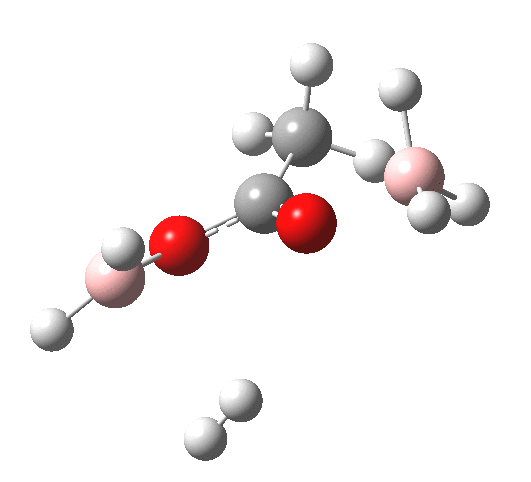
What happens if R=Me (an ester)? Well, the activation energy is now closer to 40 kcal/mol, which means the rate of the reaction would be very slow. Notice the bump corresponding to 5-coordinate boron has now vanished!

Again, a link for IRC animation of the reaction (it is rather nice, even if a say so myself). QED? Well, not quite. One still has to show that TS2 – TS4 do not control things! The IRC for TS2 (the first addition of a hydrogen to the carbon) is shown below, again with fascinating bumps along the way. The TS2 animation is here. The free energy of TS2 is 6.9 kcal/mol lower than TS1 (even though the actual activation barrier is higher), which makes the latter the rate determining step. Note the bumps at IRC = -8 and +5. These are due to rotations setting up the reaction.

TS3, a ring closing reaction (animation) shows an unexpected feature which I leave you to discover for yourself. TS4 is the second and final addition of a hydrogen to the carbon, with animation and resembling an SN2 inversion. The reaction is completed by hydrolysis.
 |
 |
The relative free energies of TS1, 2, 3 and 4 are respectively 0.0, -6.9, -35.0 and -19.4 kcal/mol, which makes the overall rate limiting step TS1. If that is the case, then this explains why borane reduces only a carboxylic acid and not an ester.
Now all I have to do is explain all of this to my tutorial group! Mind you, this is a deceptively complex mechanism, and who knows if it may yet spring surprises.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.









































